このページではFinishing関連のツールを公開しています。ツールはいずれもMacOSで動作します(20190820)
GenoFinisher、AceFileViewer、ShortReadManagerを用いると、イルミナのメイトペアデータとPCRフリーのペアエンドデータだけで多くの場合、細菌ゲノム配列を完全決定できます。
完全決定にお困りの場合はご連絡ください(連絡先 大坪嘉行 yohtsubo[at]ige.tohoku.ac.jp)。
3つのソフトウエアがあります。
 GenoFinisher ver. 2.22. (update 2020.10.26)
GenoFinisher ver. 2.22. (update 2020.10.26)
Finishingを支援するツールです。
 AceFileViewer ver. 1.65 (update 2021.03.06)
AceFileViewer ver. 1.65 (update 2021.03.06)
ACEファイルの可視化するツールです。前処理によって作成されるファイル"connectionInfo"は、GenoFinisherにロードできます。
ShortReadManager ver.1.04 (update 2020.11.26)
イルミナNGSリードを取り扱うためのツール集です。
作者が著者に含まれているもののみです。イルミナ社以外のシーケンサー由来のデータを用いたものも含みます。
- Vejarano F, Suzuki-Minakuchi C, Ohtsubo Y, Tsuda M, Okada K, Nojiri H. Complete Genome Sequence of Thalassococcus sp. Strain S3, a Marine Roseobacter Clade Member Capable of Degrading Carbazole. Microbiol Resour Announc. 2019 Jul 11;8(28). pii: e00231-19. doi: 10.1128/MRA.00231-19. PubMed PMID: 31296670; PubMed Central PMCID: PMC6624753.
- Devanadera A, Vejarano F, Zhai Y, Suzuki-Minakuchi C, Ohtsubo Y, Tsuda M, Kasai Y, Takahata Y, Okada K, Nojiri H. Complete Genome Sequence of an Anaerobic Benzene-Degrading Bacterium, Azoarcus sp. Strain DN11. Microbiol Resour Announc. 2019 Mar 14;8(11). pii: e01699-18. doi: 10.1128/MRA.01699-18. PubMed PMID: 30938329; PubMed Central PMCID: PMC6424213.
- Mpofu E, Vejarano F, Suzuki-Minakuchi C, Ohtsubo Y, Tsuda M, Chakraborty J, Nakajima M, Okada K, Tada N, Kimura T, Nojiri H. Complete Genome Sequence of Bacillus licheniformis TAB7, a Compost-Deodorizing Strain with Potential for Plant Growth Promotion. Microbiol Resour Announc. 2019 Jan 24;8(4). pii: e01659-18. doi: 10.1128/MRA.01659-18. eCollection 2019 Jan. PubMed PMID: 30701261; PubMed Central PMCID: PMC6346210.
- Vejarano F, Suzuki-Minakuchi C, Ohtsubo Y, Tsuda M, Okada K, Nojiri H. Complete Genome Sequence of the Marine Carbazole-Degrading Bacterium Erythrobacter sp. Strain KY5. Microbiol Resour Announc. 2018 Aug 30;7(8). pii: e00935-18. doi: 10.1128/MRA.00935-18. eCollection 2018 Aug. PubMed PMID: 30533913; PubMed Central PMCID: PMC6256505.
- Sugawara M, Tsukui T, Kaneko T, Ohtsubo Y, Sato S, Nagata Y, Tsuda M, Mitsui H, Minamisawa K. Complete Genome Sequence of Bradyrhizobium diazoefficiens USDA 122, a Nitrogen-Fixing Soybean Symbiont. Genome Announc. 2017 Mar 2;5(9). pii: e01743-16. doi: 10.1128/genomeA.01743-16. PubMed PMID: 28254989; PubMed Central PMCID: PMC5334596.
- Ohtsubo Y, Nonoyama S, Ogawa N, Kato H, Nagata Y, Tsuda M. Complete genome sequence of Burkholderia caribensis Bcrs1W (NBRC110739), a strain co-residing with phenanthrene degrader Mycobacterium sp. EPa45. J Biotechnol. 2016 Jun 20;228:67-68. doi: 10.1016/j.jbiotec.2016.04.042. Epub 2016 Apr 26. PubMed PMID: 27130496.
- Ohtsubo Y, Nonoyama S, Nagata Y, Numata M, Tsuchikane K, Hosoyama A, Yamazoe A, Tsuda M, Fujita N, Kawai F. Complete Genome Sequence of Sphingopyxis terrae Strain 203-1 (NBRC 111660), a Polyethylene Glycol Degrader. Genome Announc. 2016 Jun 9;4(3). pii: e00530-16. doi: 10.1128/genomeA.00530-16. PubMed PMID: 27284143; PubMed Central PMCID: PMC4901234.
- Ohtsubo Y, Nonoyama S, Nagata Y, Numata M, Tsuchikane K, Hosoyama A, Yamazoe A, Tsuda M, Fujita N, Kawai F. Complete Genome Sequence of Sphingopyxis macrogoltabida Strain 203N (NBRC 111659), a Polyethylene Glycol Degrader. Genome Announc. 2016 Jun 9;4(3). pii: e00529-16. doi: 10.1128/genomeA.00529-16. PubMed PMID: 27284142; PubMed Central PMCID: PMC4901233.
- Muraguchi Y, Kushimoto K, Ohtsubo Y, Suzuki T, Dohra H, Kimbara K, Shintani M. Complete Genome Sequence of Algoriphagus sp. Strain M8-2, Isolated from a Brackish Lake. Genome Announc. 2016 May 12;4(3). pii: e00347-16. doi: 10.1128/genomeA.00347-16. PubMed PMID: 27174266; PubMed Central PMCID: PMC4866842.
- Tabata M, Ohhata S, Kawasumi T, Nikawadori Y, Kishida K, Sato T, Ohtsubo Y, Tsuda M, Nagata Y. Complete Genome Sequence of a γ-Hexachlorocyclohexane Degrader, Sphingobium sp. Strain TKS, Isolated from a γ-Hexachlorocyclohexane-Degrading Microbial Community. Genome Announc. 2016 Apr 7;4(2). pii: e00247-16. doi: 10.1128/genomeA.00247-16. PubMed PMID: 27056231; PubMed Central PMCID: PMC4824264.
- Tabata M, Ohhata S, Nikawadori Y, Sato T, Kishida K, Ohtsubo Y, Tsuda M, Nagata Y. Complete Genome Sequence of a γ-Hexachlorocyclohexane-Degrading Bacterium, Sphingobium sp. Strain MI1205. Genome Announc. 2016 Apr 7;4(2). pii: e00246-16. doi: 10.1128/genomeA.00246-16. PubMed PMID: 27056230; PubMed Central PMCID: PMC4824263.
- Minami T, Ohtsubo Y, Anda M, Nagata Y, Tsuda M, Mitsui H, Sugawara M, Minamisawa K. Complete Genome Sequence of Methylobacterium sp. Strain AMS5, an Isolate from a Soybean Stem. Genome Announc. 2016 Mar 17;4(2). pii: e00144-16. doi: 10.1128/genomeA.00144-16. PubMed PMID: 26988053; PubMed Central PMCID: PMC4796132.
- Hirose Y, Fujisawa T, Ohtsubo Y, Katayama M, Misawa N, Wakazuki S, Shimura Y, Nakamura Y, Kawachi M, Yoshikawa H, Eki T, Kanesaki Y. Complete Genome Sequence of Cyanobacterium Leptolyngbya sp. NIES-3755. Genome Announc. 2016 Mar 17;4(2). pii: e00090-16. doi: 10.1128/genomeA.00090-16. PubMed PMID: 26988037; PubMed Central PMCID: PMC4796116.
- Hirose Y, Fujisawa T, Ohtsubo Y, Katayama M, Misawa N, Wakazuki S, Shimura Y, Nakamura Y, Kawachi M, Yoshikawa H, Eki T, Kanesaki Y. Complete genome sequence of Cyanobacterium Fischerella sp. NIES-3754, providing thermoresistant optogenetic tools. J Biotechnol. 2016 Feb 20;220:45-6. doi: 10.1016/j.jbiotec.2016.01.011. Epub 2016 Jan 16. PubMed PMID: 26784989.
- Hirose Y, Fujisawa T, Ohtsubo Y, Katayama M, Misawa N, Wakazuki S, Shimura Y, Nakamura Y, Kawachi M, Yoshikawa H, Eki T, Kanesaki Y. Complete genome sequence of cyanobacterium Nostoc sp. NIES-3756, a potentially useful strain for phytochrome-based bioengineering. J Biotechnol. 2016 Jan 20;218:51-2. doi: 10.1016/j.jbiotec.2015.12.002. Epub 2015 Dec 4. PubMed PMID: 26656223.
- Ohtsubo Y, Nagata Y, Numata M, Tsuchikane K, Hosoyama A, Yamazoe A, Tsuda M, Fujita N, Kawai F. Complete Genome Sequence of Sphingopyxis macrogoltabida Type Strain NBRC 15033, Originally Isolated as a Polyethylene Glycol Degrader. Genome Announc. 2015 Dec 10;3(6). pii: e01401-15. doi: 10.1128/genomeA.01401-15. PubMed PMID: 26659674; PubMed Central PMCID: PMC4675939.
- Ohtsubo Y, Nagata Y, Numata M, Tsuchikane K, Hosoyama A, Yamazoe A, Tsuda M, Fujita N, Kawai F. Complete Genome Sequence of a Polypropylene Glycol-Degrading Strain, Microbacterium sp. No. 7. Genome Announc. 2015 Dec 10;3(6). pii: e01400-15. doi: 10.1128/genomeA.01400-15. PubMed PMID: 26659673; PubMed Central PMCID: PMC4675938.
- Ohtsubo Y, Nagata Y, Numata M, Tsuchikane K, Hosoyama A, Yamazoe A, Tsuda M, Fujita N, Kawai F. Complete Genome Sequence of Polypropylene Glycol- and Polyethylene Glycol-Degrading Sphingopyxis macrogoltabida Strain EY-1. Genome Announc. 2015 Dec 3;3(6). pii: e01399-15. doi: 10.1128/genomeA.01399-15. PubMed PMID: 26634754; PubMed Central PMCID: PMC4669395.
- Ohtsubo Y, Moriya A, Kato H, Ogawa N, Nagata Y, Tsuda M. Complete Genome Sequence of a Phenanthrene Degrader, Burkholderia sp. HB-1 (NBRC 110738). Genome Announc. 2015 Nov 5;3(6). pii: e01283-15. doi: 10.1128/genomeA.01283-15. PubMed PMID: 26543118; PubMed Central PMCID: PMC4645203.
- Ohtsubo Y, Nagata Y, Numata M, Tsuchikane K, Hosoyama A, Yamazoe A, Tsuda M, Fujita N, Kawai F. Complete Genome Sequence of Polyvinyl Alcohol-Degrading Strain Sphingopyxis sp. 113P3 (NBRC 111507). Genome Announc. 2015 Oct 15;3(5). pii: e01169-15. doi: 10.1128/genomeA.01169-15. PubMed PMID: 26472829; PubMed Central PMCID: PMC4611681.
- Kato H, Ogawa N, Ohtsubo Y, Oshima K, Toyoda A, Mori H, Nagata Y, Kurokawa K, Hattori M, Fujiyama A, Tsuda M. Complete Genome Sequence of a Phenanthrene Degrader, Mycobacterium sp. Strain EPa45 (NBRC 110737), Isolated from a Phenanthrene-Degrading Consortium. Genome Announc. 2015 Jul 16;3(4). pii: e00782-15. doi: 10.1128/genomeA.00782-15. PubMed PMID: 26184940; PubMed Central PMCID: PMC4505128.
- Hirose Y, Katayama M, Ohtsubo Y, Misawa N, Iioka E, Suda W, Oshima K, Hanaoka M, Tanaka K, Eki T, Ikeuchi M, Kikuchi Y, Ishida M, Hattori M. Complete Genome Sequence of Cyanobacterium Geminocystis sp. Strain NIES-3708, Which Performs Type II Complementary Chromatic Acclimation. Genome Announc. 2015 May 7;3(3). pii: e00357-15. doi: 10.1128/genomeA.00357-15. PubMed PMID: 25953174; PubMed Central PMCID: PMC4424290.
- Nagao N, Hirose Y, Misawa N, Ohtsubo Y, Umekage S, Kikuchi Y. Complete Genome Sequence of Rhodovulum sulfidophilum DSM 2351, an Extracellular Nucleic Acid-Producing Bacterium. Genome Announc. 2015 Apr 30;3(2). pii: e00388-15. doi: 10.1128/genomeA.00388-15. PubMed PMID: 25931606; PubMed Central PMCID: PMC4417702.
- Hirose Y, Katayama M, Ohtsubo Y, Misawa N, Iioka E, Suda W, Oshima K, Hanaoka M, Tanaka K, Eki T, Ikeuchi M, Kikuchi Y, Ishida M, Hattori M. Complete Genome Sequence of Cyanobacterium Geminocystis sp. Strain NIES-3709, Which Harbors a Phycoerythrin-Rich Phycobilisome. Genome Announc. 2015 Apr 30;3(2). pii: e00385-15. doi: 10.1128/genomeA.00385-15. PubMed PMID: 25931605; PubMed Central PMCID: PMC4417701.
- Miura T, Uchino Y, Tsuchikane K, Ohtsubo Y, Ohji S, Hosoyama A, Ito M, Takahata Y, Yamazoe A, Suzuki K, Fujita N. Complete Genome Sequences of Sulfurospirillum Strains UCH001 and UCH003 Isolated from Groundwater in Japan. Genome Announc. 2015 Mar 26;3(2). pii: e00236-15. doi: 10.1128/genomeA.00236-15. PubMed PMID: 25814615; PubMed Central PMCID: PMC4384155.
- Ohtsubo Y, Kishida K, Sato T, Tabata M, Kawasumi T, Ogura Y, Hayashi T, Tsuda M, Nagata Y. Complete Genome Sequence of Pseudomonas sp. Strain TKP, Isolated from a γ-Hexachlorocyclohexane-Degrading Mixed Culture. Genome Announc. 2014 Jan 30;2(1). pii: e01241-13. doi: 10.1128/genomeA.01241-13. PubMed PMID: 24482516; PubMed Central PMCID: PMC3907731.
- Shintani M, Ohtsubo Y, Fukuda K, Hosoyama A, Ohji S, Yamazoe A, Fujita N, Nagata Y, Tsuda M, Hatta T, Kimbara K. Complete Genome Sequence of the Thermophilic Polychlorinated Biphenyl Degrader Geobacillus sp. Strain JF8 (NBRC 109937). Genome Announc. 2014 Jan 23;2(1). pii: e01213-13. doi: 10.1128/genomeA.01213-13. PubMed PMID: 24459274; PubMed Central PMCID: PMC3900906.
- Ohtsubo Y, Sato T, Kishida K, Tabata M, Ogura Y, Hayashi T, Tsuda M, Nagata Y. Complete Genome Sequence of Pseudomonas aeruginosa MTB-1, Isolated from a Microbial Community Enriched by the Technical Formulation of Hexachlorocyclohexane. Genome Announc. 2014 Jan 23;2(1). pii: e01130-13. doi: 10.1128/genomeA.01130-13. PubMed PMID: 24459257; PubMed Central PMCID: PMC3900889.
- Ohtsubo Y, Fujita N, Nagata Y, Tsuda M, Iwasaki T, Hatta T. Complete Genome Sequence of Ralstonia pickettii DTP0602, a 2,4,6-Trichlorophenol Degrader. Genome Announc. 2013 Oct 31;1(6). pii: e00903-13. doi: 10.1128/genomeA.00903-13. PubMed PMID: 24179121; PubMed Central PMCID: PMC3814637.
- Minegishi K, Aikawa C, Furukawa A, Watanabe T, Nakano T, Ogura Y, Ohtsubo Y, Kurokawa K, Hayashi T, Maruyama F, Nakagawa I, Eishi Y. Complete Genome Sequence of a Propionibacterium acnes Isolate from a Sarcoidosis Patient. Genome Announc. 2013 Jan;1(1). pii: e00016-12. doi: 10.1128/genomeA.00016-12. Epub 2013 Jan 15. PubMed PMID: 23405284; PubMed Central PMCID: PMC3556827.
- Ohtsubo Y, Maruyama F, Mitsui H, Nagata Y, Tsuda M. Complete genome sequence of Acidovorax sp. strain KKS102, a polychlorinated-biphenyl degrader. J Bacteriol. 2012 Dec;194(24):6970-1. doi: 10.1128/JB.01848-12. PubMed PMID: 23209225; PubMed Central PMCID: PMC3510582.
NGSの登場によってドラフトゲノム配列を手に入れることは容易になりました。 PacBioシーケンサーなどを使えば、環状ゲノムを構成でき、「高精度に決定できた」と言われます。 この精度は、100株のゲノム配列をざっくり決定してバイオインフォマティクス的に知見を発見するには満足のいく精度かもしれません。 ところが1つの細菌株を深く研究する場合にはこの「高精度」は必ずしも十分満足のいく精度ではないことがしばしあることと思います。
精度99.99%では、10 kbに1つエラーがあることになり、ホモポリマーサイトでの塩基数間違いが多いPacBioなどの場合、遺伝子おそよ10個に1つが、 pseudo遺伝子であると間違えてアノテーションされることになります。
細菌のゲノム配列を高精度(1塩基も間違えないレベルで)に完全決定するのには様々な課題があります。 GenoFinisherシリーズのソフトウエアには、これら課題を解決するために作成した多くのツールが含まれています。
これらツールを使用すると、多くの場合、イルミナ社のショートリードデータだけを用いて、インシリコでの完全決定が可能です。 PCRフリーのペアエンドデータとメイトペアデータがあることが理想です。 「必ずできる」という訳ではありませんが、経験上90% (n = 45)程度は追加実験なしで決定が可能です。インシリコだけで完全決定が不可能な場合でも、数カ所のgapあるいは正確に決定できない部位が残るだけのケースが多いです(ほぼ同じリピート配列が50コピー以上あったりこれらが複数セットあると難航します)。
十分に慣れた場合の作業時間は、最も簡単なゲノムの場合で3時間、一般的なゲノムで10時間程度、少々困難なもので2週間程度です。
このページでは、GenoFinisher、AceFileViewer、ShortReadManagerを用いた細菌ゲノム配列の完全決定法について、概略を説明します。もし、PCRフリーのペアエンドデータとメイトペアデータをお持ちで、手助けが必要でしたらご連絡ください(共同研究としていただけると幸いです)。
イルミナシーケンサーでデータを取得してください。PCRフリーのペアエンドデータとメイトペアデータが必要です。どちらも100x 程度の量があると良いです。 アセンブルにリードを過剰に投入するとかえってアセンブルの質が低下するので、実際にはリードデータの一部だけを使うことがよくあります。
SRMのfindmateツールを使って、ゲノム中でお互いに向かい合った向きの関係になるように、メイトペアリードを処理します。
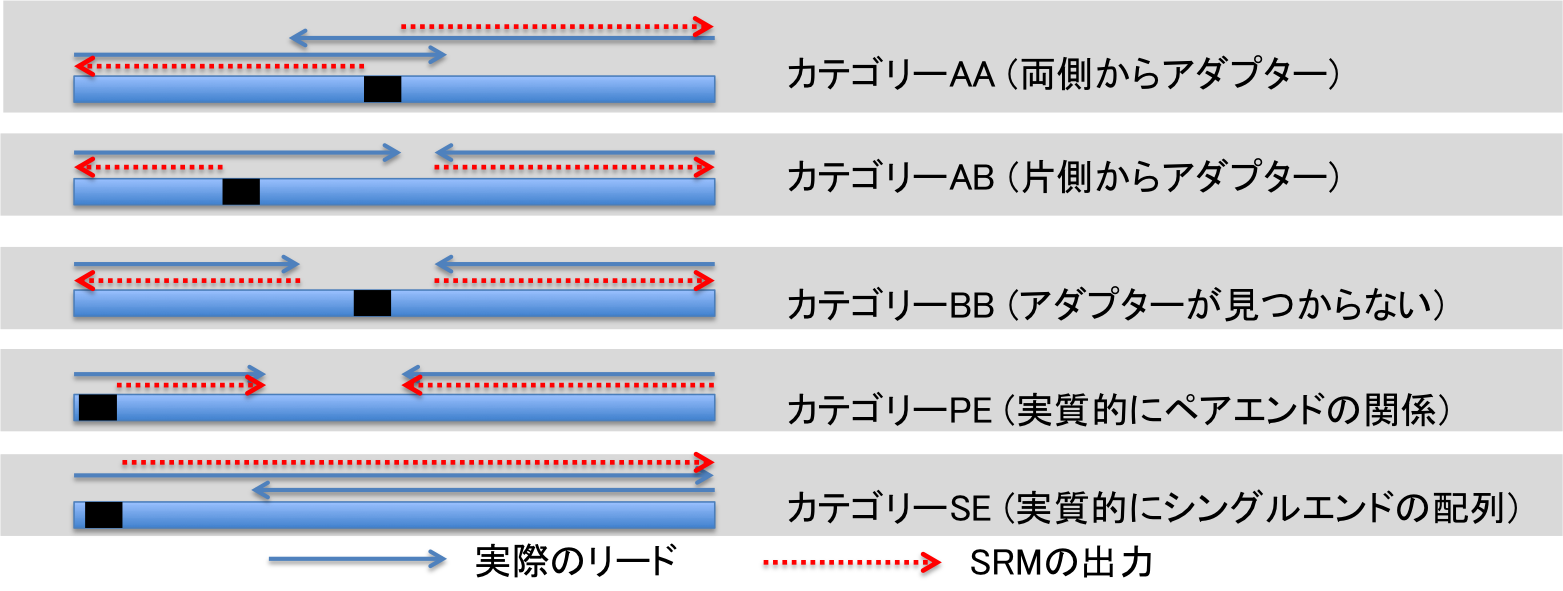
findmateツールはリード中にアダプター部分(ビオチンラベルされていた38 bpのアダプター)がどのように検出されたに基づいて、いくつかのカテゴリーに分類して(異なるファイルに)リードを出力します(図1)。 メイトペアデータの末端間距離はそれぞれ異なる分布に従うので、AA、AB、BBいずれかのカテゴリーのものだけで十分量あると理想的です。
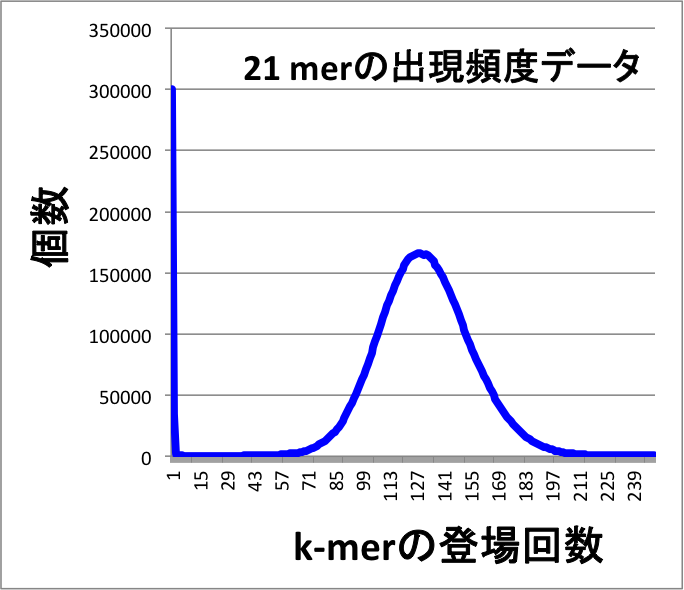
SRMを使用して、k-merの登場回数に基づいたリードのトリミングを行います。 まず、全リードデータ(findmate済みのリードとPCRフリーのイルミナリードデータを合わせると良い)についてk-merの登場回数の分布を調べます(図2)。登場回数が低いk-merは、シーケンスエラー、あるいは、アダプターDNAとゲノムDNAのジャンクション部分など、偶発的に生じたk-merであると考えられ除去します。この処理を行ってからアセンブルを行うと、早く、良いアセンブルが得られます。
Newblerによりアセンブルします。version 2.8を勧めています。(1) single aceファイルを出力する(single ACE fileにチェック)、(2)0塩基以上の長さの(つまり全部の)コンティグの配列を出力する(all contig thresholdを0に)、設定が必須です。minimum read lengthは45とすると良いと思います。
AFVを起動し、出力されたaceファイルを選択して、前処理を実行します。この処理により、(1) どのコンティグとどのコンティグが何本のメイトペアリードで、どのように関連づけられていたのかのデータ、 (2)いつくかの複数のコンティグに帰属するリードについて、どのコンティグにどのように帰属しているかについてのデータ、 が収集されファイル「connectioninfo」に出力されます。収取されたデータはGFでの描画に用いることができます。
GFで、newblerの出力フォルダを選択して、runボタンを押します。計算処理がすぐに一時停止し、この時、AFVから出力されたconnectioninfoファイルを指定します。コンティグをGUI上で並べ(自動でも並べられます)、どのようなアセンブルであるか把握します(図3)。
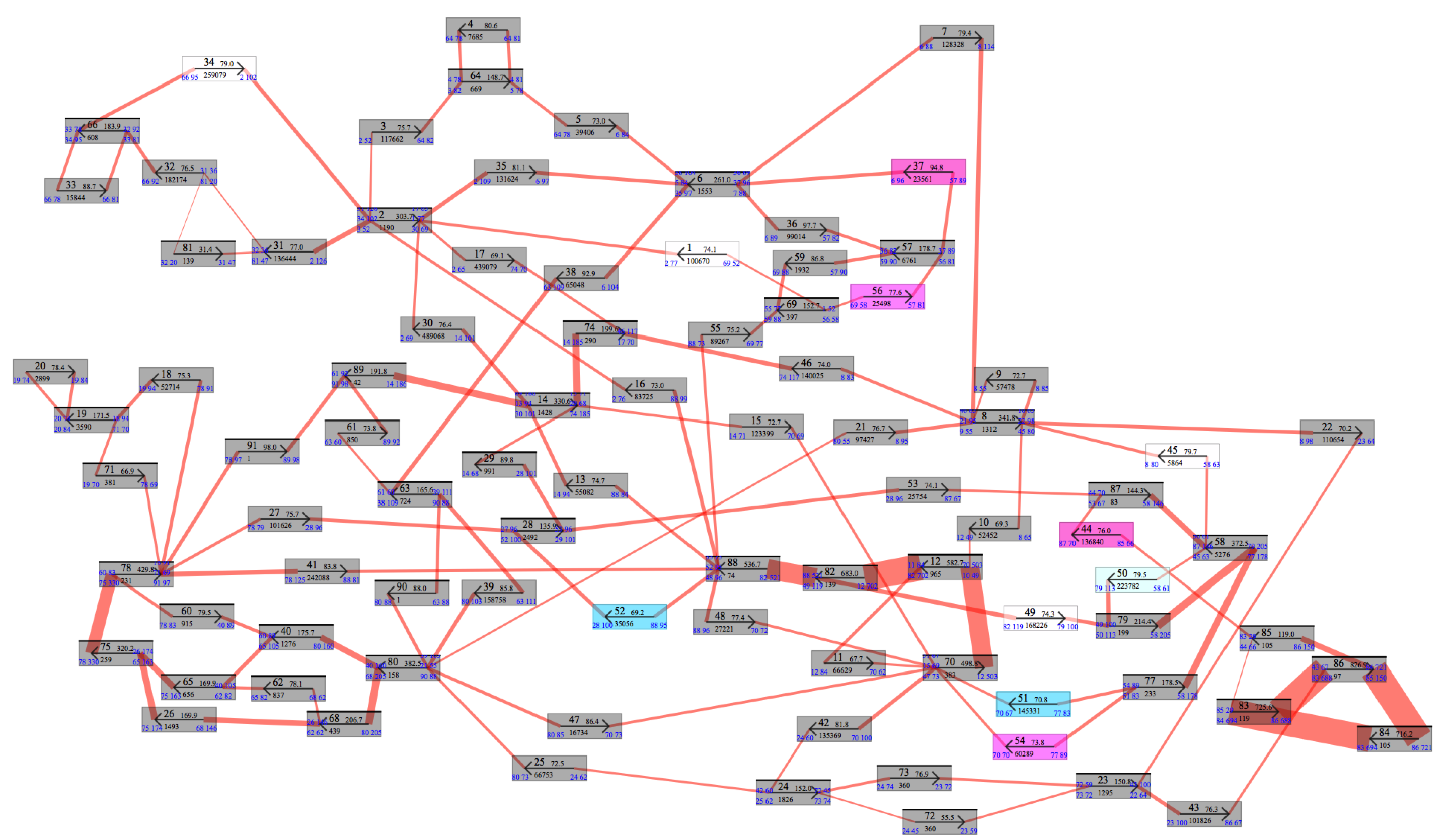
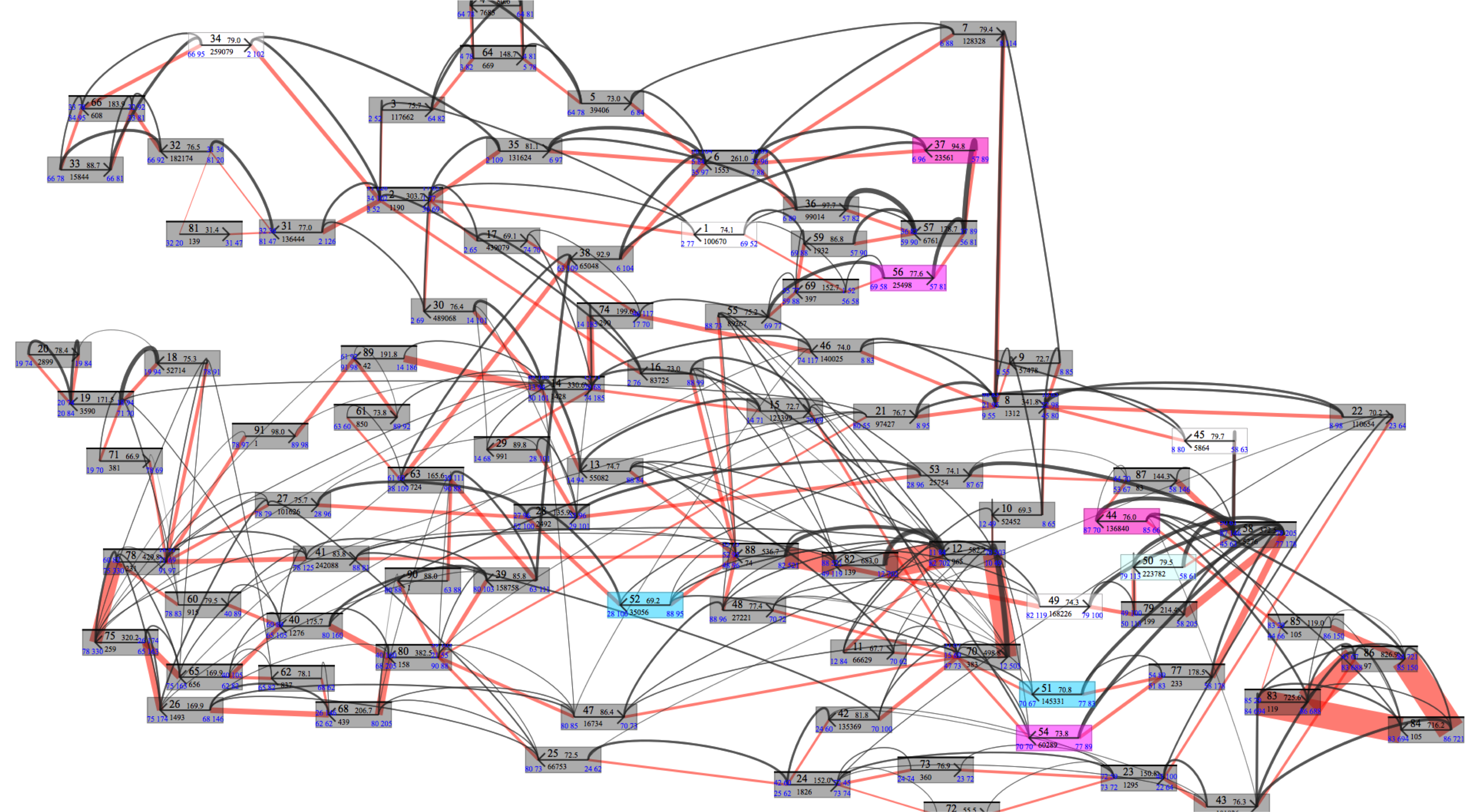
図4は、connectioninfoファイルにあったコンティグ間が何本のメイトペアでブリッジされていたかを図3に追加表示したところです。ここでは全て表示していますのでごちゃごちゃしていますが、特定のコンティグについてだけ表示させることもできます。 (1)メイトペアデータなどを参考にコンティグの繋がり方を全て明らかにし、(2)重複コンティグの配列をコンテクスト依存的に決め、(3)gapがあればその部分の配列を決定し、(4)実際に配列を貼り合わせたら、ゲノム配列が決定できたことになります。
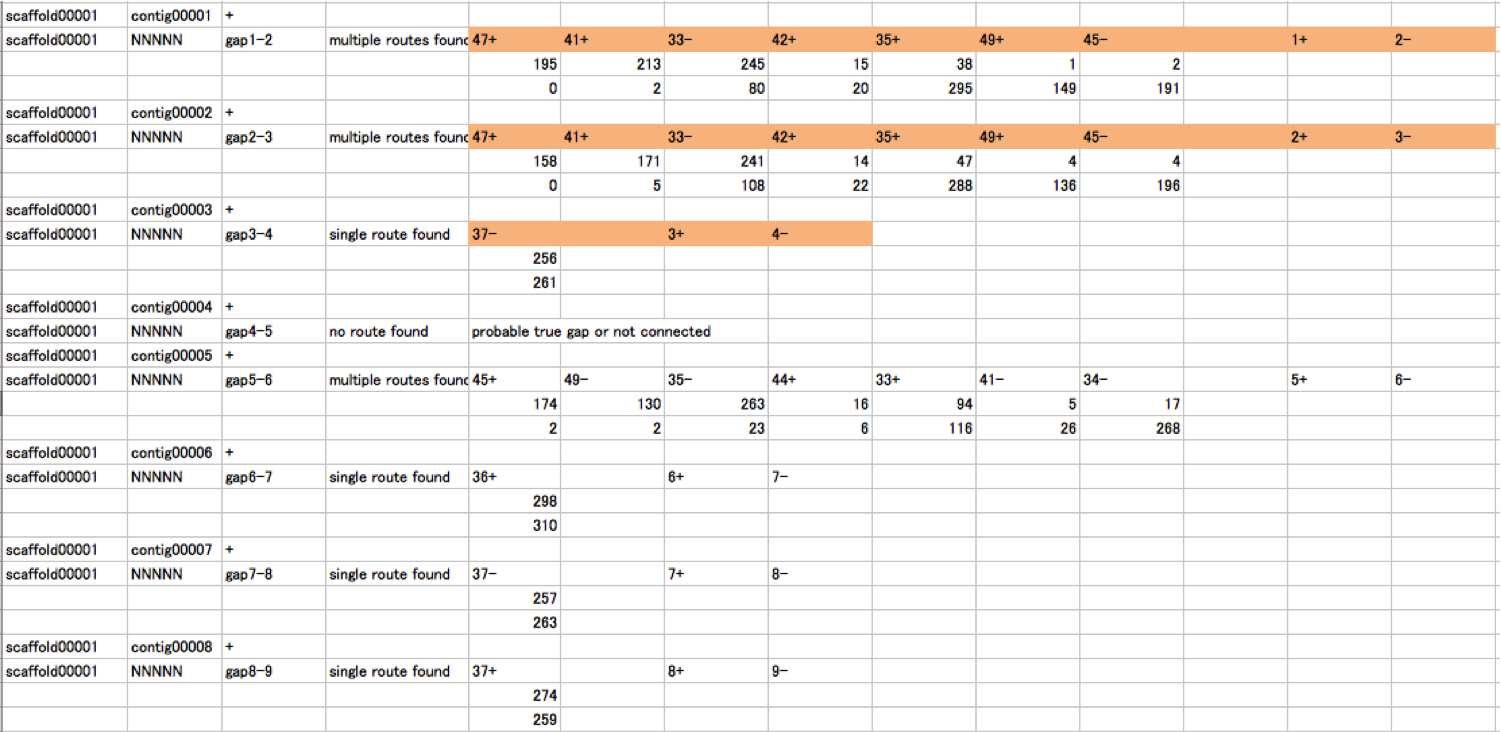
またGFから出力されたテキストデータを表計算シートに貼り付けます。この表計算シートに貼り付けたデータは、フィニッシング作業を効率良く行うために重要なデータとなります。このシートの上で作業を進めることをお勧めします。 このシートでは(図5)、スキャフォールド中の各gapについて、どのようなルートがあり売るのか、またgap前後のコンティグと、gapコンティグをブリッジするメイトペアデータが何本あるかを表示できます。このデータは、そのコンティグを並べて見てルートをチェックするのに有用で、さらには、AFVでコンテクストを踏まえてリピート配列を決定するのにも使うことができます。

図6は、図5と同じエクセルシートを、もう少し広い範囲で示したものです。以下に述べる様に、このルートの妥当性をGFで判定し、良いとなれば、AFVでの正確な配列決定を行うことになります。 配列が決定できたらエクセルシート中のNNNNNのところを実際に決定できた配列に置き換え、先頭列が空白である行を消去すると、GFの配列を張り合わせる機能にそのまま利用できる形式になります。
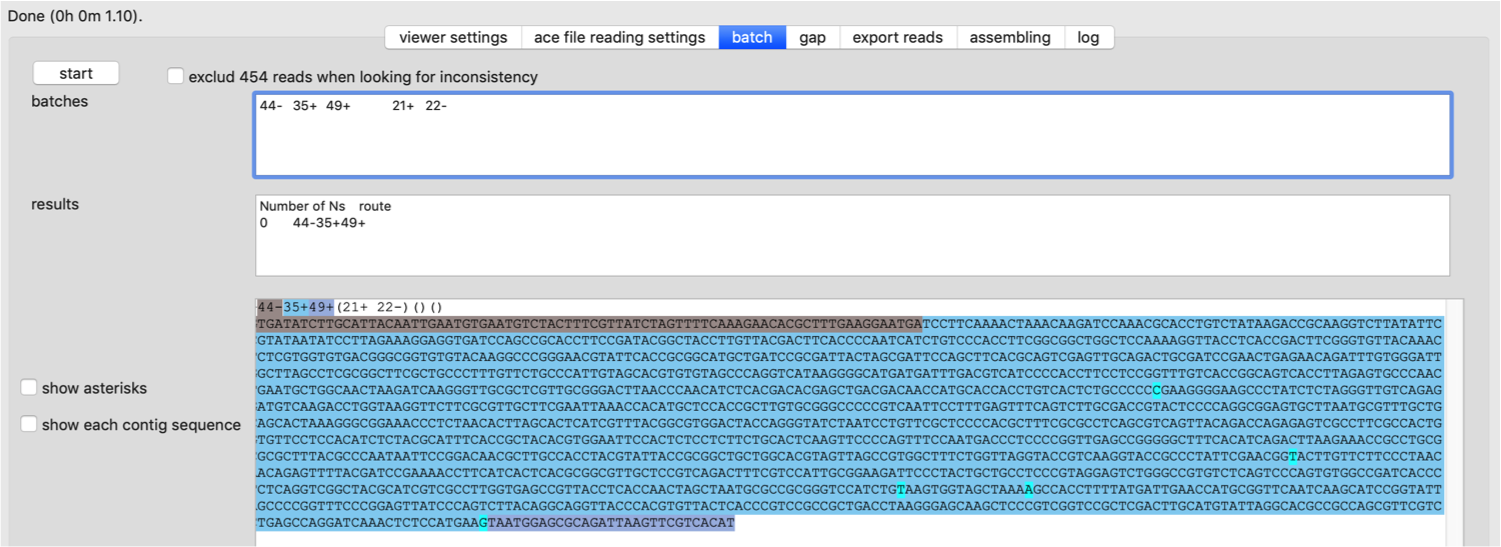
ここではギャップのクロージングの方法について述べます。配列の貼り合わせはGFの機能でできますので(後述)、その時に必要な形式となるように表計算シート上でデータをまとめます。
ギャップはスキャフォールド間のGAP(大文字で書きます)と、スキャフォールド内のgap(小文字で書きます)に大別できます。GAPはメイトペアデータがブリッジできないような長いリピート配列(例えばribosomal RNAオペロン)に起因します。 gapはリピート配列に起因するもの(repeat gapと呼びます)、gap前後の配列が本当はオーバーラップしているのにそれを切り分けてしまったことによるgap(overlap gapと呼びます)、あるいはリードが足りないために生じるgap(true gapと呼びます)、gapがあると思わせて実は存在しないgap(pseudo gap)があります。
GAPの配列決定: あるスキャフォールド末端から別のスキャフォールド末端へのコンティグルートをGFを使って探します。ほとんどの場合、ルートが見つかります。時に、ルート上のリピートコンティグの中にあるバリエーション部位にマップされているリードの相方がどのコンティグに属しているかを利用します。 ルートが見つかったら、AFVを利用して実際の配列を決定します。

repeat gapの配列決定
先のエクセルシートのデータに、スキャフォールド内のあるコンティグ末端から別のコンティグ末端へのルートの候補が(場合によってはいくつか)出力されています。 このルートが適切であるがGF上で確認します。 OKとなれば、AFVでリピートコンティグについてコンテクスト(前後のコンティグ)を指定して配列を決定します。
overlap gapの配列決定
末端がオーバーラップしている関係にあるコンティグ末端がしばしばあります。 リードの厚みが十分ないため、あるいは、このgap部分を通過するリードがないため、newblerが切り分けてしまった場合に生じます。 まずはAFVを使用して、末端がオーバラップしているかどうかを調べます(図)。 (1)末端がどのように重複しているかを調べた出力図、 (2)gapがオーバーラップしていると仮定した場合に、末端部分をブリッジするメイトペアデータの末端間距離分布が異常でないかどうか、 (3)リファレンスゲノム中でこの部分がどうなっているかを調べた図、 を参考にします。またアセンブルに使用しなかったリードの中に、この部分の配列を含むものがあるかどうかSRMを使って探し出し、 見つかればSeqViewを使うなどして、overlapしているとして良いか判定します。
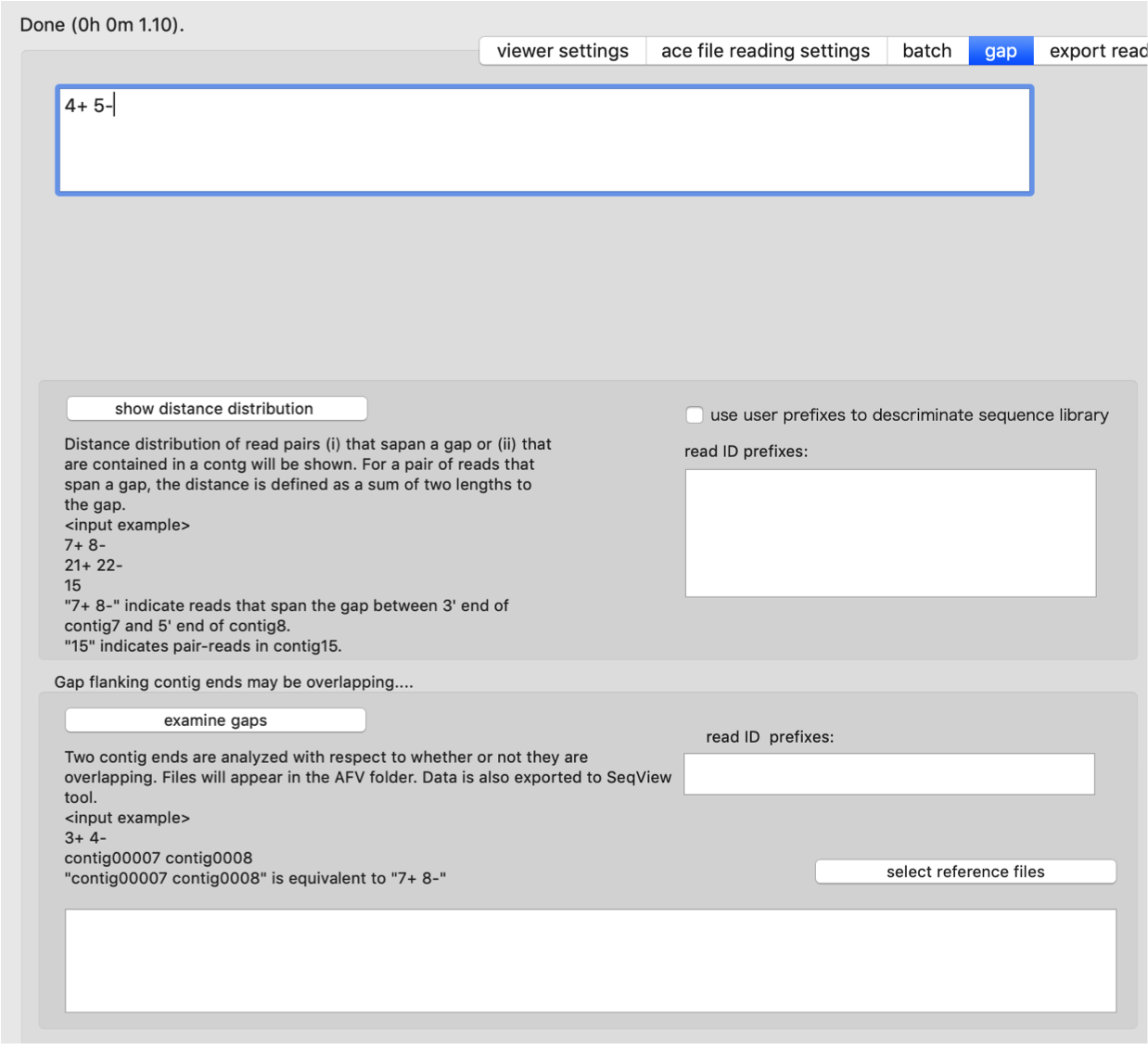
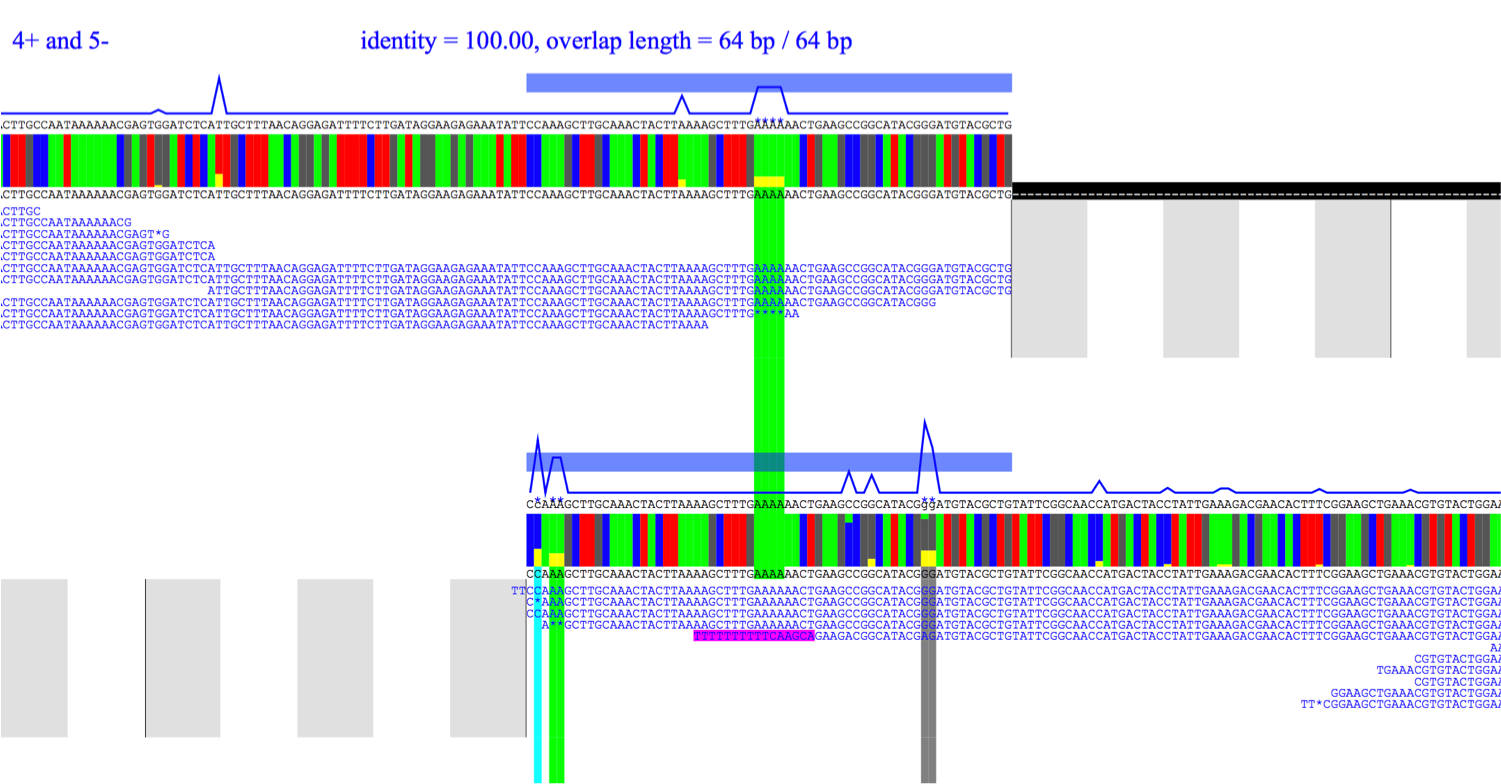
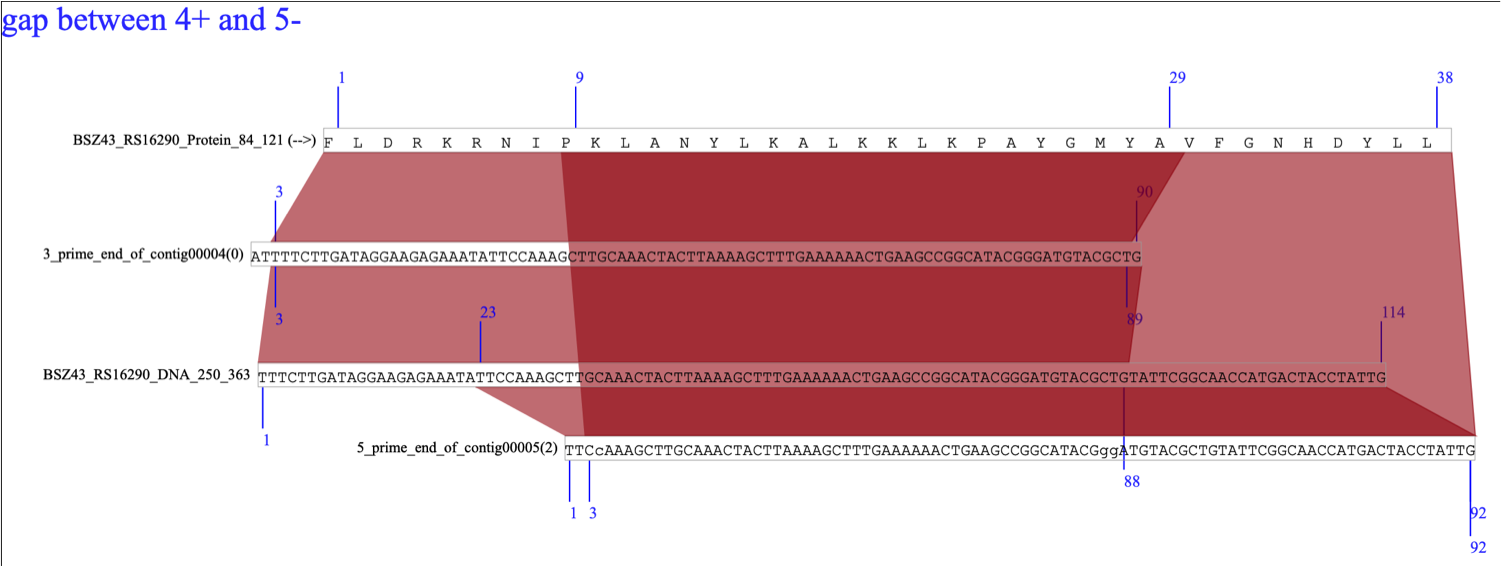
true gapの配列決定
末端がオーバーラップしていない場合、リードがないためのgapであると考えられます。 アセンブルに使わなかったリードがあれば、その中からgapに関連するリードをSRMを使って抽出します。 SRMでは、配列を指定して、FASTQあるいはmulti FASTAファイルから、その配列を含むリードを抽出できます。 検索には、指定した配列に相補的な配列を含めることもできます。また、見つかった配列のペアとなるリードを出力することもできます。 gapを埋めるようなリードが見つからなければ、gapがどれくらいの大きさであるかを、gap部分をブリッジするペアリードの末端間距離分布を調べるなどして検討をつけ、PCR増幅して配列を決定します。
実はgapではないgap(pseudo gap)の同定
GFでコンティググラフを見るとコンティグ間にgapは存在しないことがわかることがあります。 さらにAFVでその部分を解析し、実際にどうであるか判定します。 このようなgapはイルミナシーケンサーが配列特異的に、非ランダムに読み取りエラーを起こすためと思われます。
overlap gapが見出されている場合、オーバラップ部分の配列を削ります。手作業は間違いを犯しやすいので、GFのコマンドライン様機能を利用してコマンド入力でオーバラップを一括除去します。
コマンドでは例えば「delete contig=4 end=3 length=8 seq=CCAAAGCT」のように用います。間違えて2度以上削除してしまわないように、配列を指定するようにしてあります。コマンドは複数同時にコピーペーストで入力できます。
GFの配列連結機能を使用します。GFでは、コンティグ名と向き、gapにつけた名前と配列、をそれぞれ複数指定(表計算シートで編集)して一括して配列を連結することができます。できた配列には新たに任意の名前を設定するようにします。この新たにつけた名前は、さらに次の連結に使うことができます。 この機能を使って、GAPに名前をつけ、コンティグとgap配列を貼り合わせてGAP配列を作成します。またコンティグとgap配列を指定してスキャフォールドを作成します。作成したスキャフォールドやGAPをさらに貼り合わせてゲノム配列とします。

GFのFinishCheckerで配列をチェックします。FinishCheckerでは2通りのアルゴリズムでチェックが可能です。 (1)k-merを利用した方法: 元々の配列データには存在しないのになぜか完成した配列に存在する様なk-mer (例えば21-mer)があれば、 その部分は何らかの間違いがある可能性があります。コンティグの向きを間違えて貼り合わせてしまった場合、セレクトオールあるいはコピーしようとしてAあるいはCが挿入されてしまった場合などのミスを見出せます。 (2)メイトペアデータ使った方法: メイトペアデータを完成配列にマッピングし、異常な部分がないか観察します。ペアのうち一方しかマップされないリードが局在している部分、ゲノムの各点をブリッジするペアの末端間距離分布が他と比べて異常な部分、ペアの位置関係あるいは距離が異常であるものが局在する部分、などに着目します。 おかしな箇所があれば、どこをどう間違えたのか明らかにし、必要に応じて修正して、再度チェックします。おかしなところが見られなくなったら完成です。 FinishCheckerを利用するにはGFのメイン画面で連結によって作製したスキャフォールドを右クリックしてからFinishCheckerを選択してください。
細菌ゲノムには、ゲノムによって300塩基から600塩基程度ですが、特定の配列が5から20回程度タンデムにリピートしている部分がある場合があります。これらが全部完全に同じ配列であれば良いですが、リピートの中に多少のバリエーション部位がある場合がほとんどです。このような箇所についてバリエーション部位まで含めて正確に決定するのはとても時間がかかりますが、多くの場合決定可能です。簡単に説明すると以下の方法が有効です(必ずうまくいくかどうかはわかりません)。このような部位は、コンティグ数個がループを形成している様に見えます。(1)各コンティグについて、バリエーション部位について調べ、何通りの配列が、それぞれ何コピーずつあるか、調べます(AFV)。 各配列パターンに名前をつけます(例えばcontig50a、contig50b、contig50c....)。ループの外側のコンティグのループ側末端に着目して、AFVで解析し、ループの開始部分がどのコンティグのどのパターンであるか決定します。 例えば20+_50a_51b_52c_______50a_51a_52a_21+のようになっていることがわかります。次に、リピートコンティグのバリエーションのうち、コピー数が1であるものに着目し、その前後のコンティグのバリエーションが何であるかを決めます。 例えば、50bが1コピーであるとして、その前後が51c-52b-50b-51a-52dである、というようにです。このようにして、これらコンティグの全部のバリエーションに付いて並びを矛盾無く説明できれば、配列が決定できたことになります。