ここではGenomeMatcherのメイン機能について説明します。このページで使われている基礎的な用語についてはこちらを、GenomeMatcher特有の言葉や設計コンセプトについてはこちらをご覧ください。
概要
GenomeMatcherの主要機能では、2つの配列についてその大規模構造から微細構造まで幅広いスケールでの比較結果の可視化が可能です。生成された比較イメージ図が貯留されていつでも呼び出せること、比較イメージの任意部位をドラッグすることで次の解析の比較範囲とできること、などで使い勝手の向上がはかられています。
- シーケンスの入力方法
- 配列の比較プログラム
- 比較範囲の変更/指定
- グラフィックス出力形式
- SNPの表示
- アラインメントの出力
- カテネーション(連結) mode
- mesh mode
- synteny analysis
- colorgram
- XY mode
- Location lookup
- 保存の仕方
- DotMatch
- 注意とヒント
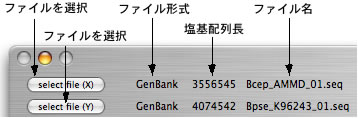
読み込めるファイル形式についてはこちらをどうぞ。ここではmulti FASTA形式は受け付けません。読み込むとファイル形式、塩基配列長などが表示されます。
いくつかの配列比較プログラムによる比較結果を、グラフィカルに出力できます。利用可能な比較プログラムは以下の通りです。実行上の制約についてはこちら。
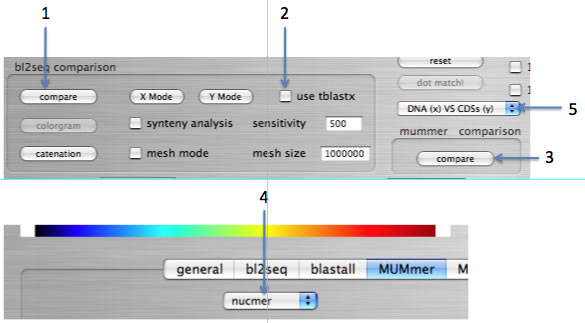
bl2seqは「bl2seq comparison」ボックス内のcompareボタン(下図1)から実行します。bl2seqのtbalstxを実行するには、当該box内のuse tblastxチェックボックス(下図2)をオンにします。bl2seqのblastnを実行するには、これをオフにします。
MUMmerボタンは 「mummer comparison」ボックス内のcompare(下図3)ボタンから実行します。nucmerを使うには環境設定のMUMmerタブ内のプルダウンメニュー(下図4)でnucmerを、promerを使うにはpromerを選択します。
blastallによる比較は、プルダウンメニュー(下図5)からプログラムを選択することで実行します。CDSs VS CDSsを選ぶとblastpが、それ以外を選ぶとtblastnが実行されます。なおCDSs (x) VS DNA (y)では配列XのCDSをクエリ、配列Yをデータベースとして、blast検索が実行されます。
また比較結果をベクターグラフィックスに書き出すには、「イメージ」メニューから対応するメニューを選択します。選択後に行われる解析の結果がPDFファイルに保存されます。
配列比較プログラムで比較する範囲は、自由に変更できます。変更するにはいくつか方法があります。
- すでに比較イメージがある場合、イメージの上をドラッグする。ドラッグ範囲が解析範囲指定テキストボックスに入力されます。
- 解析範囲指定テキストボックスに、解析範囲を手入力する。
- ナビゲーションボタンをクリックする(平行移動します。移動距離はstep sizeで指定します)。
- すでに比較イメージがある場合、イメージの上をシフトを押しながらドラッグする。
- 比較範囲を、配列の開始から終了にセットするにはresetボタンを押します。
- 貯留イメージをクリックすると、ドラッグなどによる解析範囲の変更がリセットされます。
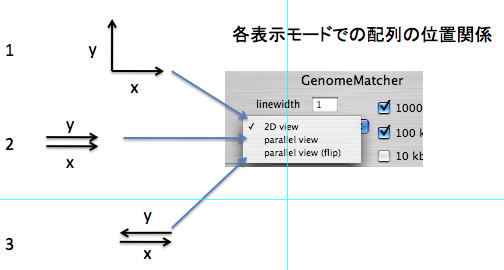
大きく分けて2つのグラフィックス出力形式があります。
- 左下を原点とした直交座標系でのグラフィック出力(下図1)
- x軸に読み込んだ配列を下側、y軸に読み込んだ配列を上側に置いたグラフィック出力(下図2,3)
後者に関しては、y軸に読み込んだ配列のposition 1が左側にくる置き方(下図2)と、右側に来る置き方(下図3)があります。この出力形式の場合、ナビゲーションボタンの配置位置等が変わる、ドラッグによる範囲選択の仕様が変わる、blastallのblastpで比較した際にidentity scoreが表示される、などいくつかの変化があります。
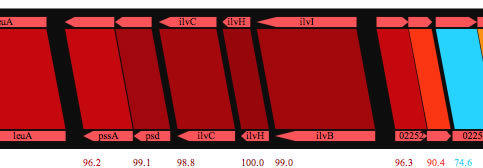
2の出力の例。数字はblastpによるidentityスコア。
blastの結果をテキストファイルでお持ちの場合、GenomeMatcherを利用して比較イメージを描画することができます (version 1.415以降)。結果ファイルは、bl2seqにオプション-Dに0を指定して実行することで得られる形式でなくてはなりません。以下に適切なファイル形式について示します。
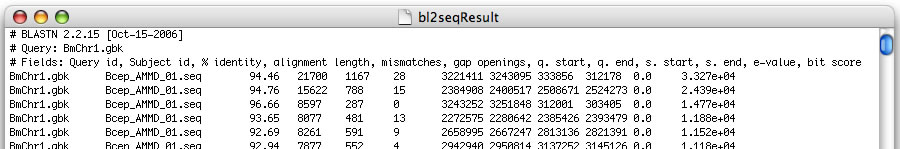
このように、bl2seqにオプション-Dに0を指定して実行すると最初の3行は相同性に関するデータではありません。blastallのblastnの結果を描画させる時は、最初の3行が無視されないように適当な空行を挿入してください(\nで改行してください)。ファイルからロードしたデータに基づいて描画するには、「use loaded data」チェックボックスをオンにしてください。
- ロードしたデータによる描画は、通常と同様、PDFファイルに書き出すことができます(コマンド+G)。
- ロードしたデータがtblastx由来のデータである場合は、use tblastxチェックボックスをオンにしてください(カラースケールがアミノ酸配列用に切り替わるだけです)。
- meshモードと併用しないでください。
- 解析範囲を指定して、描画することもできます。
MUMmerのnucmerで解析した場合、検出されたSNPの違いを表示できます。極度に相同性の高い配列の中のどこに違いがあるかをグラフィカルに表示するときなどに便利です。indelを青で、transitionを白で、transversionを緑で表示します。(transition はA→G、G→A、C→T、T→C、transversion はそれ以外の置換)
各種プログラムによる配列のアラインメント結果を出力できます。メイン画面中の「show alignments by」ボックス内のプルダウンメニューからプログラムを選択してください。利用できるプログラムは以下の通りです。実行上の制約についてはこちら。
- bl2seqのblastn
- bl2seqのtblastx
- clustalW
- MAFFT
- MUMmerのnucmer
- MUMmerのpromer
bl2seqおよびMUMmerについては、どちらのプログラムを使うかの設定方法はこちらで述べた通りです。clustalWとMAFFTに関しては、clustalW-reverseあるいはMAFFT-reverseを選択すると、x軸の配列とy軸の配列の相補鎖のアラインメントが出力されます。
複数あるレプリコンやゲノムを相互に比較するための機能です。例えばそれぞれ3つのレプリコンを持つ10個のゲノム(全部で30レプリコン)について総当たりでの比較が可能です。以下の手順に従ってください。
- メイン画面で「catenation」ボタンを押して、専用のwindowを開きます。
- レプリコンを含むファイルパスを編集します。add filesボタンをクリックして同じゲノムに属するレプリコンのファイルを全て選択してください。
- ゲノムが他にもある場合は、同様の操作を繰り返します。
- 以上の操作により、テキストフィールドに沢山のファイルパスが並ぶことになります。このとき、同じゲノムに属するレプリコンへのファイルパスは同じ行に入ります。複数のゲノムを指定した場合は、複数行にわたってファイルパスが表示されます。
 概念的にはファイルパスはこのように並びます。
概念的にはファイルパスはこのように並びます。
- 「create and set concatenated sequence」ボタンを押します。これによりファイルを指定した順に、そこに含まれるDNA配列が連結されます。連結配列はメイン画面にセットされます。また、各レプリコンのサイズ、各ゲノムのサイズが専用window左側にあるテキストボックスに表示されます。
 レプリコンのサイズはこのように並びます。
レプリコンのサイズはこのように並びます。 ゲノムのサイズ(各レプリコンサイズの総和)がこのように並びます。
ゲノムのサイズ(各レプリコンサイズの総和)がこのように並びます。
- この操作を、X軸、Y軸の両方について行います。
- レプリコンおよびゲノムの境界に引く線の色と太さを指定します。
- 「TIFF image to main window」ボタンを押すか、「PDF image to file」ボタンを押します。前者では、メイン画面に比較結果が描画されます。後者ではPDFファイルに画像が出力されます。この際、描画サイズを変更することができます。ポスターなどに幅一杯に描画したいときなどにご利用下さい。
この機能の注意点は以下の通りです。
- 各レプリコンについて配列の全長を比較することになります。
- 全部の配列が内部的に連結されてから比較されます。各塩基の位置番号は最初のレプリコンからの通し位置番号となります(1つめのレプリコンが1000bpのとき、2つめのレプリコンの1塩基目は、連結配列では1001塩基目となります)。
bl2seqでは10Mb程度の配列であれば、(メモリ上)問題なく解析を終えることができます。しかし100Mbの配列は解析の途中でクラッシュしてしまいます。またbl2seqのtblastxであれば1Mb程度の配列の比較であっても途中でクラッシュしてしまいます。これはbl2seqの実行時にメモリが足りなくなるためのようです。GenomeMatcherでは長い配列であっても比較が可能なように、配列を一定の大きさごとに区切り、部分の比較を繰り返すことで全体の比較をする機能がついています。この機能を使うにはメイン画面の「bl2seq comparison」ボックス内のmesh modeチェックボックスをオンにし、どれぐらいの大きさに区切るか(mesh sizeと呼びます)を指定して下さい。この機能はsynteny analysisとは併用できません。またbl2seqの解析のみが実行できます。
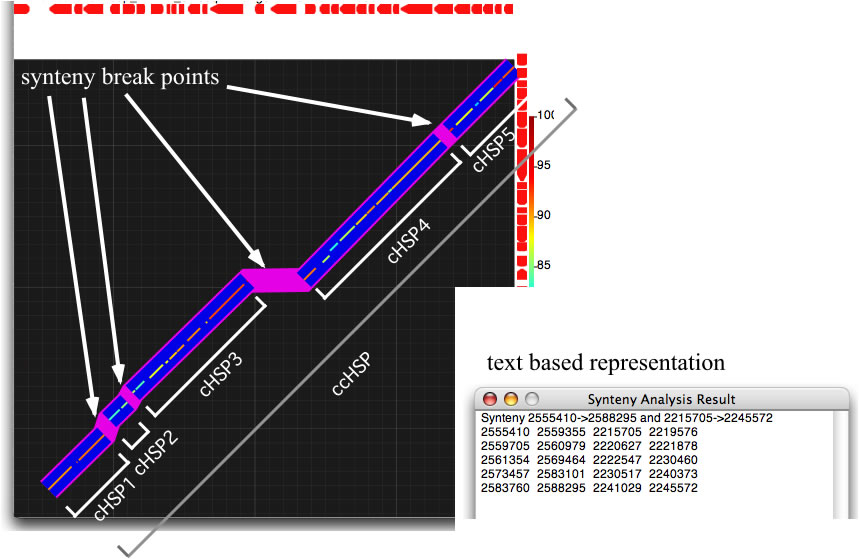
外来領域が挿入されるとシンテニー(遺伝子の並び)に不連続が生じます。逆に不連続な場所(ギャップ)を探すと、そこには外来領域があるかもしれません。bl2seqのblastnではグラフィックス上断続的な線として現れるHSPが得られます。しかし外来領域を同定する上で意味の無いような小さいギャップが多く存在しているため、それなりの大きさを持った外来領域を同定するのは大変です。GenomeMatcherのsyntensy analysisは、小さいギャップを埋めることで、大きなギャップの同定を支援します。
メイン画面の「bl2seq comparison」ボックス内のシンテニー解析のスイッチがオンの時にbl2seqのblastnで解析すると、お互いに近い位置(sensitivityで指定します)にあるHSP同士が連結されます。ここでは複数のHSPが連結されたものをcHSP(connected HSP)と呼びます。cHSPsは太い青い線で示されます。さらに複数のcHSPsは、ある値( 固定値:20kb)をしきい値として連結されます。連結されたcHSPをccHSP(connected cHSP)と呼びます。ccHSPsは紫色の折れ線で示されます。 また結果はテキスト形式でも出力されます。テキスト形式では、ccHSPの開始座標および終了座標に始まり、ccHSPを構成するいくつかのcHSPの開始座標と終了座標が出力されます。すなわち比較的大きなギャップの位置をここから取り出すことができます。sensitivityの単位はbaseです。500と指定してあると500base以内のギャップがあるHSP同士を連結します。
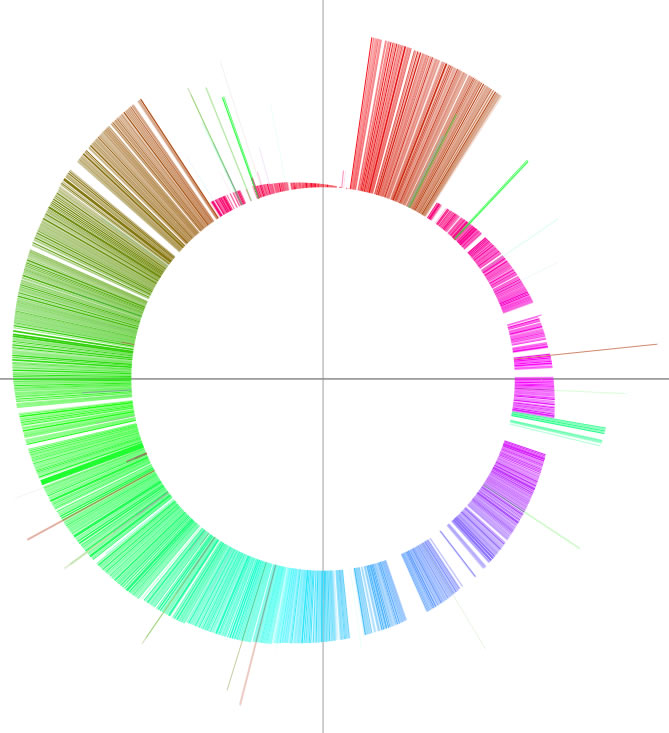
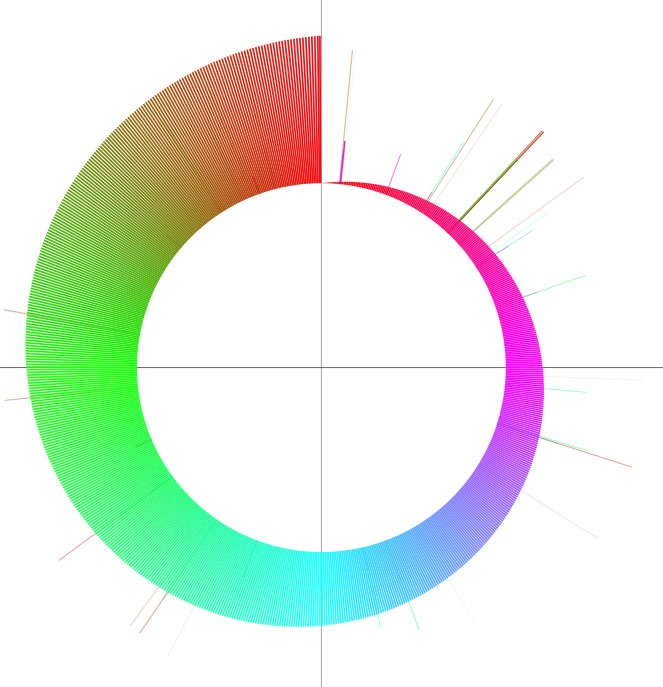
2つのゲノムの形の違いを表現するための機能です。2つの配列の全長が解析範囲になっている状態でcolorgramとかかれたボタンをクリックしてください。配列Xの各部分が、配列Yのどの部位に似ているかが表示されます。配列Xは0時から時計回りに配置されています。カラフルな線がたくさん現れると思いますが、線の「高さ」と「色」は両方とも配列Yのどの位置に相同配列が存在するか、を表しています。一度の逆位によって形状の違いの説明がつく2つのゲノムの比較を例として示します(左)。またある配列をそれ自身と比較した結果も併せて示します(右)。描画ウィンドウ上のテキストボックス中の値を変えることで絵の見た目を調整することができます。また右側にあるテキストボックスに特定の形式に従ってデータを入力することで、任意の位置に直線と文字列を追加することができます。
DNA配列を比較した時に得られるイメージの中でお互いに離れた2つの部分を近づけて見るための機能です。2つの部分は垂直方向あるいは水平方向のいずれかにのみ離れていなくてはなりません。この機能は染色体の再編成部位の末端領域に重複した部分があるかどうか、またどのような遺伝子が末端に位置しているか、などを調べるための機能です。本機能で使える比較プログラムはbl2seqのblastnおよびtblastxのみです。「synteny analysis」および「mesh mode」との併用はできません。利用するには、メイン画面の「bl2seq comparison」ボックス内の「X Mode」ボタンあるいは「Y Mode」ボタンを押してください。
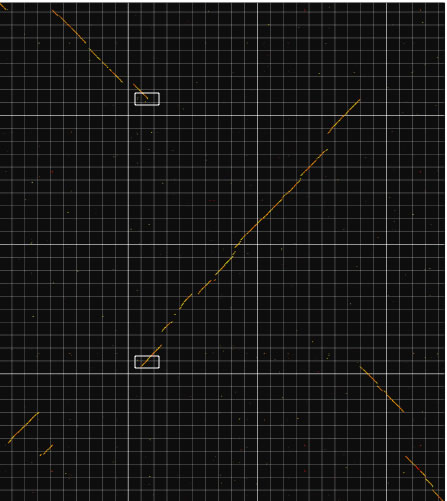
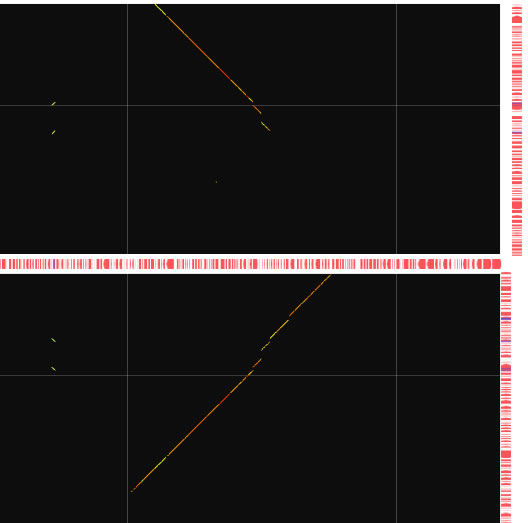
Y modeの実行例。垂直方向に離れた2つの部分(左)を、近くに表示することができます(右)。ここでは50kb超の重複部分が、染色体の再編成部位になっていることを見ることができます。
配列Xおよび配列Yに対して各種の検索を行う機能があります(コマンド+Lでウィンドウが開きます)。プルダウンメニュー中の5つの検索機能から1つを選んで「excecute」ボタンを押すと、配列Xおよび配列Yの両方に対して検索が実行されます。
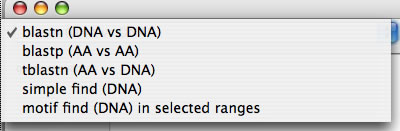
上から順に、
- blastn検索:配列全体に対してbl2seqのblastnがかかります。クエリは1件のみ指定できます。
- blastp検索:配列全体に対してblastallのblastpがかかります。クエリは1件のみ指定できます。
- tblastn検索:配列全体に対してbl2seqのtblastnがかかります。クエリは1件のみ指定できます。
- simple find検索:配列全体に対して100%マッチする配列を、配列の表と裏の両方に対して検索します。クエリは複数件指定できます。
- motif find検索:配列の選択部分に対してある程度のミスマッチを許容して似ている配列を探します。クエリは複数件指定できます。
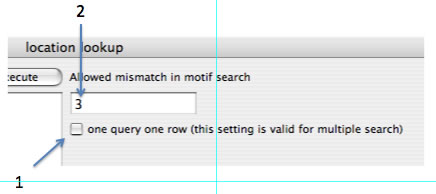
後ろ2つの機能については、出力形式を変更することができます(右側にあるチェックボックス(下図1)をオンにしてみてください)。またmotif find検索をするときは何baseのミスマッチを許容するかを、右側のテキストボックス(下図2)で指定してください。motif findは非常に時間がかかりますので、短い範囲を指定しておおよその時間をつかんでから実行するようにしてください。
同時に複数の配列を検索できますので、DNA配列に対して何度も検索をしなくてはいけないとき等に便利です。
イメージカラムを保存することができます(「ファイル」メニューの中の「イメージカラムを保存」)。たくさんのイメージがあるとファイルサイズがやたらと大きくなってしまいます。イメージカラムの中の絵が数枚になるように、不必要なイメージを消去してから保存してください。
「dot match」ボタンを押すと、別の解析システムが起動します。dot matchでは2つの配列間の相同性について、例えば2塩基以上のマッチを全て検出して表示します。何塩基以上のマッチを検出するかについては2通りの設定が可能で、例えば3塩基以上のマッチを黒で、10塩基以上のマッチを赤で、表示することができます。使用上のヒントは以下の通りです。
- 解析範囲は常に正方形です。2つの配列は常に直交座標系に配置されます。
- 2つの配列それぞれについて、解析開始位置を指定します。解析終了位置は、開始位置に解析長を加えることで自動的に算出されます(解析終了位置を直接していすることはできません)。
- 表示中のwindowのコピーを1つだけ作ることができます。
- メイン画面の方で、注釈を入力するとこちらにも反映されます。デザインしたprimerのアニーリング位置、挿入配列の末端など、アノテーションとして追加することで、これらの位置を見ながら解析ができます。
- 一辺の長さが100bp以下になると塩基がグラフィックスに沿って表示されます。
- コマンド+Zでundo(取り消し)ができます。
- ドラッグにより解析範囲を変更できます。
- 挿入配列の末端の同定、転写ターミネーターの同定、CRISPRの同定などに使えます。この際XとY両方に同じ配列を読み込むことをおすすめします。
- PCR増幅がうまくいかないときに、dotmatchで当該領域を解析すると、高次構造を取り得る部分が見つかることがあります。
- あまりに短い相同領域は、目で見えない場合があります。線を太くすると見えるかもしれません。ただし、線の太さと長さを勘違いしないように気をつけて下さい。
- 背景を黒くすると印刷時にトナー代がかさむかもしれません。「white backgournd」をオンにするとトナーを節約できます。
- イメージカラムのイメージを消去するには、コマンド+Dを押します。
- メイン機能では、設定を変更してもそれが反映されるのは次の解析からです。例えばカラースケールを変更しても、現在表示されているイメージの色調は変わりません。この点が付属機能「Compare sequences」とは異なります。
- ヒントの表示/非表示の設定は環境設定のgeneralタブでできます(ソフトの再起動後に反映されます)。
- 翻訳コード(translation table)を変更できます(環境設定のgeneralタブ)。
- tblastxを実行するとあまり意味のない線が沢山表示されるかもしれません。e-valueをきつめに設定するか、環境設定でidentity score thresholdを高めに設定すると改善します。