GenomeMatcherのご利用ありがとうございます。本取扱説明書を読むまえにいくつかの点にご留意下さい。
- GenomeMatcherの画面上にあるパーツの多くには、利用上のヒントが書き込まれています。ポインタを重ねて下さい。
- このソフトウエアでどのような解析が可能なのかについて知りたい人はこちらのギャラリーをご覧ください。
- 動画マニュアルを参照できますここからどうぞ。
- ソフトウエアは最新バージョンをダウンロードの上ご利用下さい。最新バージョンについてはここをクリックしてください。
- GenomeMatcherはMacOS10.3以降で動作します。解析に要する時間はスペックに強く依存します。
- Q and Aを用意しています。
- エラーが起きるときは気を付けて欲しい操作、してはいけない操作、既知のバグ、についてご覧ください(将来的に問題解決の予定です)。バグの報告を歓迎します。連絡先までご報告下さい。
- 同じテストサンプルを使って「Compare」ボタンを押してから画像が表示されるまでの時間を比較してみました( n = 1 )。テスト配列のサイズはは3.45Mbと3.56Mbです。
| time (秒) | OS | クロック周波数 | プロセッサ名 | メモリ |
| 3.6 | 10.4.11 | 2 x 3 GHz | Dual-Core Intel Xeon | 4 GB |
| 7.5 | 10.3.9 | 2 x 2.5 GHz | Power PC G5 | 2 GB |
| 9.3 | 10.3.9 | 2 x 1.8 GHz | Power PC 970 | 2.5GB |
| 11.7 | 10.3.9 | 1 x 1GHz | Power PC G4 | 512 Mb |
| 16.5 | 10.3.9 | 1 x 933MHz | Power PC G4 | 640 Mb |
GenomeMatcherの概要
GenomeMatcherは2つの塩基配列間で相同性が見られる領域をディスプレイ上で2次元にカラフルに描画するソフトウエアです。DNA配列のどの部位が、どれぐらい似ているのかについて簡単に知ることができます。GenomeMatcherは基本的に既存のDNA配列比較プログラムのグラフィカルユーザーインターフェースです。DNA配列比較プログラムとしては、主にBLASTプログラムの一種であるbl2seqを利用します。「bl2seqのblastnによる配列比較結果の描画機能を基本としていろいろな機能を追加したものがGenomeMatcherである」とご理解いただくと、システム構成が理解しやすいと思います。
比較プログラムとしてはbl2seqに加えて、既存の比較プログラムであるMUMmer、MAFFT、ClustalW、に加えオリジナルのdotmatchと呼ぶアルゴリズムを用いることが出来ます。このうち、グラフィカルな結果出力を得ることができるのはbl2seq、MUMmer、dotmatchです。bl2seq、MUMmerによるグラフィカルな出力として、2つの配列を平行に配置した描画と、垂直に配置した描画の両方を描くことができます。アラインメントは、bl2seq、MUMmer、MAFFT、ClustalWによるものを出力できます。
生成された比較イメージは、その上をドラッグすることで次の解析の対象となる塩基配列の部位を指定することができます。対象範囲を指定した後は、お望みのプログラムを使って再解析できます。選択して再解析、という操作を繰り返すことで、任意の部位について詳しく調べることができます。
描画された比較イメージの周囲には、注釈情報の存在を示す矢印が表示され、これをクリックすることにより注釈情報を参照することができます。このことによりユーザーは、配列の違いをアノテーション情報と関連づけて理解することができます。
GenomeMatcherでは、非常に長い塩基配列を、ユーザが指定した長さで区切った領域ごとに比較することを何度も繰り返すことで全体の比較イメージを構築することが出来ます。この指定長は一定値とすることも出来ますが、catenationモードと呼ぶ動作モードでは、複数の指定長をセットすることが出来ます。例えばcatenationモードでは、マルチレプリコンよりなる細菌ゲノム10個を連結した塩基配列について、レプリコンごとに比較することが出来ます。
GenomeMatcherは、「遺伝子群のスケール通りの描画」にも用いることができます。表計算ソフトを使ってアノテーションデータを編集して入力し、その絵を描くことができます。描画した絵はベクトル形式で保存が可能なので、グラフィックソフトを用いた改変が容易です。
バージョン1.280から1.331への変更点は以下の通りです。
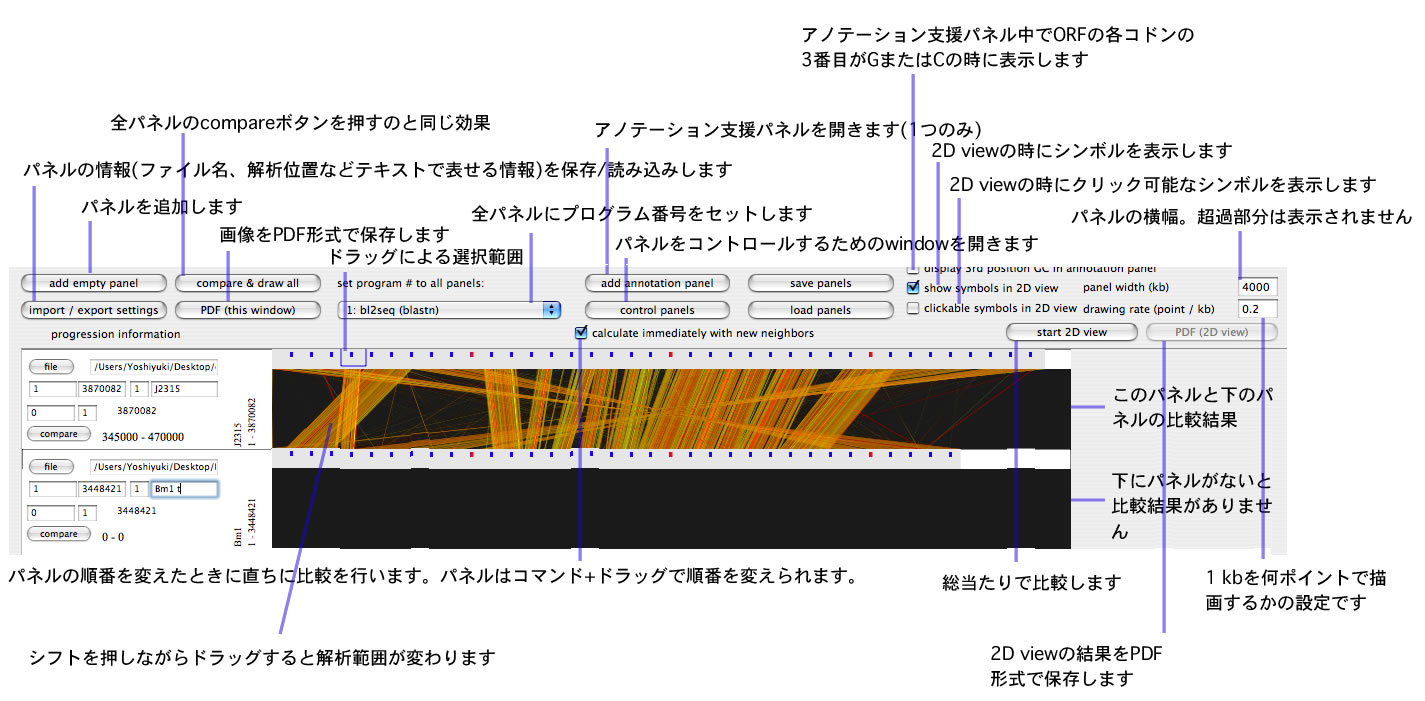
- 配列Aを配列Bと比較、配列Bと配列Cを比較、というように1対1の比較を繰り返して実行するモードを付けました<実行例>(コマンド+2でwindowが開く)。配列は基本的に何本でも読み込めます。
- 1対1の比較モードから、多対多の比較を実行できます。
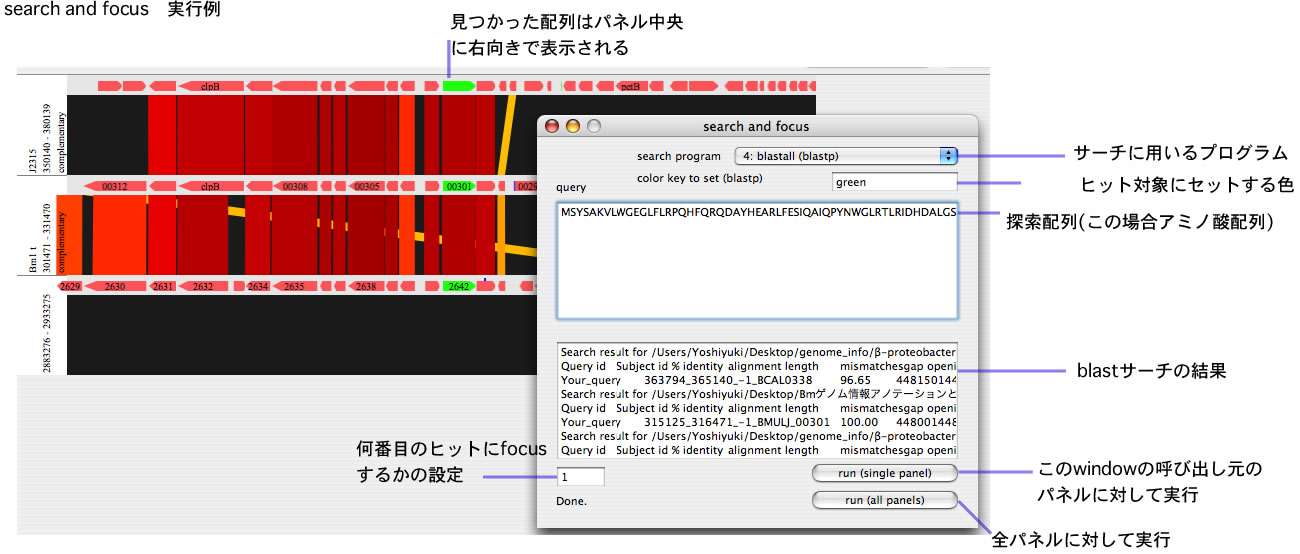
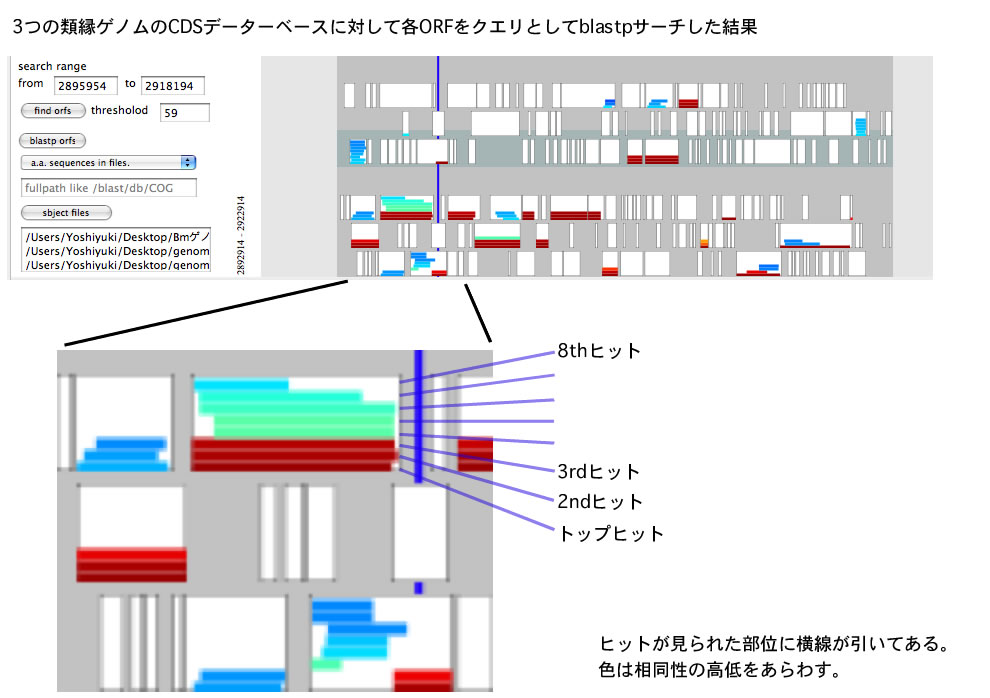
- サーチ&フォーカス機能を使うと、興味ある遺伝子の存在領域を解析範囲にすることができます。例えば5つのゲノム配列を読み込んでおき、シグマ70のアミノ酸配列でサーチ&フォーカスすると、各パネルの中央にシグマ70をコードする遺伝子が右向きにくるように解析範囲がセットされ、比較が実行されます。トップヒットにフォーカスしますが、2nd ヒット、3rd ヒットにフォーカスすることもできます。またこのときヒット対象に、指定した色を付けることができます<実行例>。
- シフトキーを押しながらパネルをドラッグすると解析範囲を変更できます。
- 遺伝子に任意の色を付けておき、その遺伝子と相同性のある遺伝子に同じ色を付ける機能があります。各塩基配列中の相同遺伝子に同じ色をつけることができます。遺伝子一つ一つについて実行することもできますが、あるパネルに含まれる遺伝子すべてについて一度に実行することもできます。
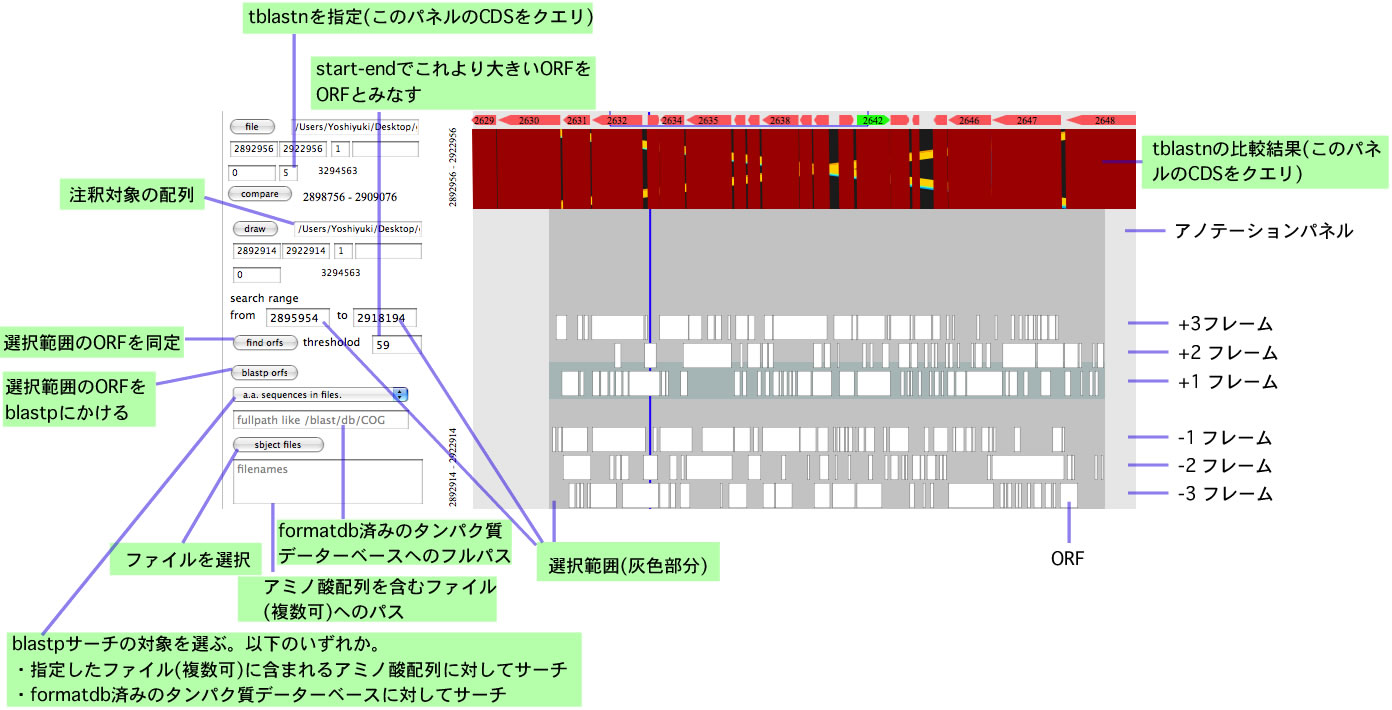
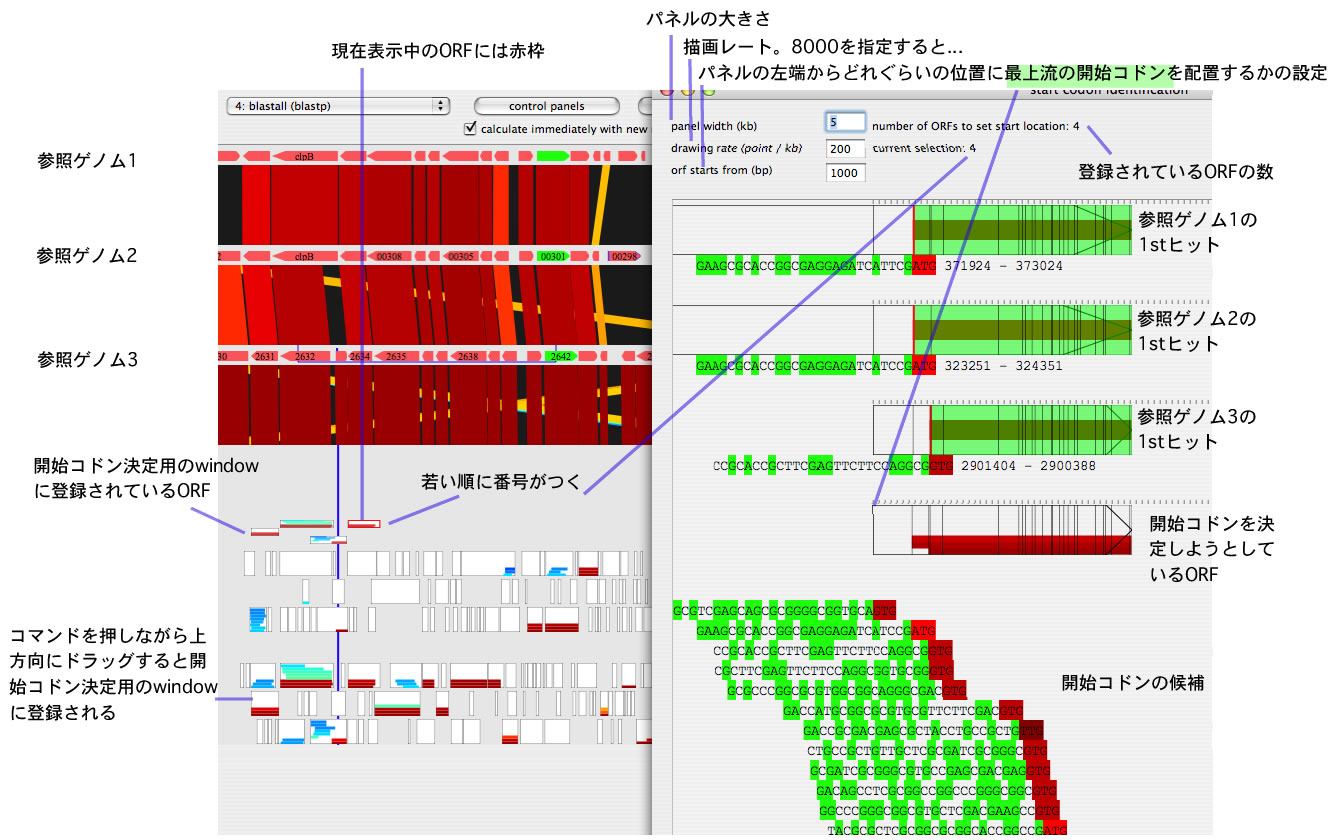
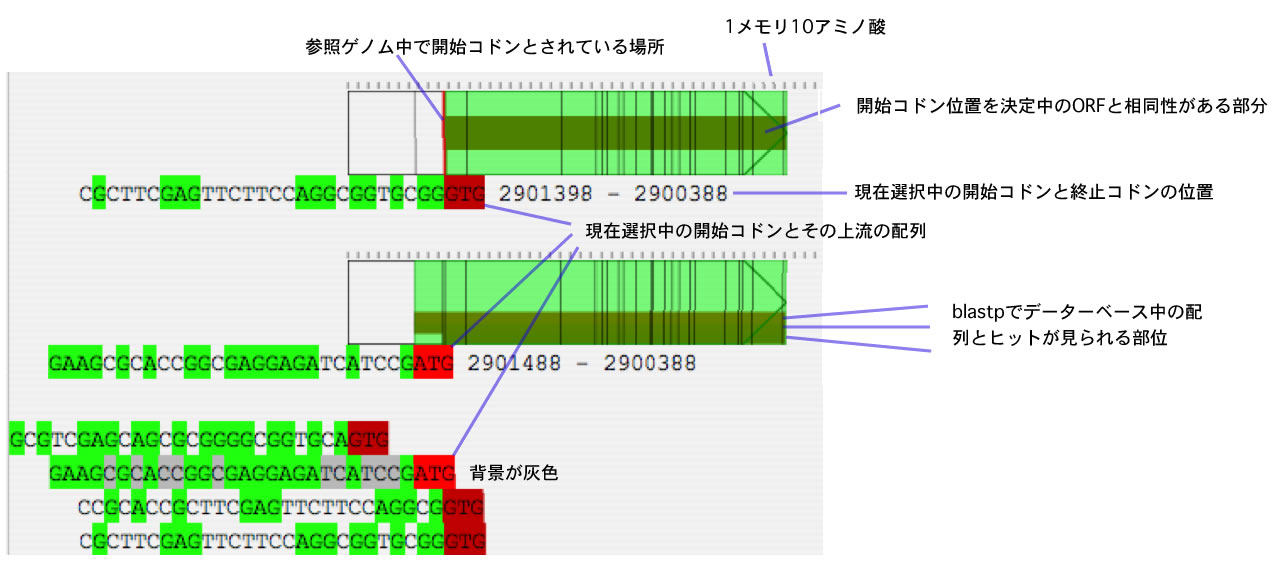
- アノテーション支援機能を付けました。ORFからCDSを選び出すステップと開始コドンの位置を決定するステップを支援します。前者については抽出したORFを任意のデーターベースに対してblastpにかけた結果がORFの上にグラフィカルに表示されますので<実行例>どのORFがCDSであるかを判定するのが容易です。後者については、複数のリファレンスゲノムそれぞれについてもっとも相同性の高いCDSについて、どの開始コドンが採用されているかについて開始コドンとその上流の塩基配列を見ることができます。また、CDSのどの部位に相同性が検出されるかも見ることができます<実行例>。
- 大量の塩基配列データについてblastデータベースに対して検索を行い、設定値以上のヒットがみられる配列を抜き出す機能を追加しました(MassiveBLAST、コマンド+3)。次世代シーケンサーによって大量に産生された配列から、相同性に基づいて配列を抽出することができます。実行例: およそ5000本よりなる16SrDNAのデータセットに対して長さ60bp-75bpの400万本の配列をblastnにかけ30分程度(2 x 3 GHz、Dual-Core Intel Xeon)で16S rDNAと相同性のある1500本程度を抽出できます。
バージョン1.25から1.270、1.280への変更点は以下の通りです。
- 環境設定画面の一部がタブブラウズとなりました。
- ツールチップの表示、非表示を設定できるようにしました(アプリケーションの再起動後に有効)。
- MUMmerによる比較結果、 blastpあるいはtblastnによる比較結果を、PDF形式で保存できるようにしました。
- 相同性についてしきい値を設定出来るようにしました。つまりある程度以上の相同性があるものだけについて 表示できます。相同性のスコア分布を見ながら(コマンド+B)、どの辺にしきい値を設定すればより見やすくなるかを判断してください。
- blastpによるCDS間の比較結果を図示できるようにしました<実行例>。x軸にあるCDSおよびy軸にあるCDSについてblastpで比較し、ヒットがあれば円で示します。相同性の高低は、カラースケールに従って色調で表示されます。またこの機能に関して、クエリの全長の何%より長い長さのヒットがあったかによって、表示、非表示を設定できるようにしました。また得られた相同性スコアについても何%より高い物を表示するかについて設定できるようにしました(コマンド+Bで開く設定画面で設定)。
- 上の機能に関連して、x軸にあるCDSをクエリにしてy軸にあるDNAについてtblastnで比較した結果を表示する機能を付けました。 y軸にあるCDSをクエリとしてx軸にあるDNAについてtblastn出来るようにしました(バージョン1.280)。
- 付属機能として、"SequenceRetriever"を付けました。x軸あるいはy軸のDNA配列から、位置と向きを指定して、DNA配列あるいはアミノ酸配列を取得することが出来る機能です。位置と向きは複数を同時に指定できます。
- 付属機能として、 "StringFormatter"を付けました。文字列を処理するための機能です。例えば、文字列を1文字ずつタブで区切る機能、 表計算シートで、各行に"配列の名前"<タブ>"配列"がデータとしてある場合、これをFASTA形式に変換する機能<実行例>、連続する2つ以上のスペースを1つに変換する機能、指定可能な2種類のタグで挟まれた文字列を指定の文字列に変換する機能、改行キー(¥nから¥rあるいはその逆)を変更する機能などがあります。
- 付属機能として"ExtractFromGenBankFile"を付けました。GenBank形式のファイルに含まれる注釈情報は、表計算シートで 解析するのに適していません。この機能は、GenBank形式のファイルに含まれる注釈情報を抽出し、表計算シートでの処理に向いた形に整形します。GenBank形式のファイルを指定すると、このファイルで使用されているFeatureKeyおよびQualifierが表示されます。抽出したいFeatureKeyおよびQualifier以外を消去し、Qualifierについては抽出したい順番に並べ替えてください。実行を押すと、表計算シートでの処理がしやすい形式に変更されます。1つのFeatureKeyについて2つ以上のnoteクオリファイアが設定されていることがありますが、何件目まで抽出するかを指定することができます(最大でいくつのnoteクオリファイアが一つのエントリにあるかについては自動で解析されます。変更しなければ全件が抽出されます)。位置情報について、joinまたはorderが使用されている場合は、最小値、および最大値が抽出された上で、CAUTION列にもともとの位置情報文字列が表示されます。
- 付属機能として"BLASTInterFace"を付けました。ローカルでBLASTを実行する機能です。ファイル中のあるいはテキストフィールドに貼り付けた問い合わせ配列を、作成済みのblast databaseに対して、あるいは指定したファイルから作成されるdatabaseに対して、BLAST検索を行うことができます。BLASTの結果は、通常の実行結果として出力されるだけでなく、通常の実行結果を表計算シートで管理するのにより適した形式に変更して出力することができます。
- 付属機能として"DinucleotideBiasSignature"を付けました。DNA配列の塩基組成には偏りがあることが知られています。 あるDNA配列について二塩基の出現頻度(AA、AC、AG、・・・TT)を調べたとき、それぞれの二塩基について出現頻度の偏りを計算することが出来ます(16個の小数値データが得られます)。この16個の小数値よりなるデータを"bias data"と呼びます。あるDNA配列Aと、別のDNA配列B(これはAの一部分かも知れません)それぞれについてbias dataが計算できますが、この差をbias differenceと呼びます(16個の小数値それぞれについての差の絶対値の総和)。 この付属機能では、ファイル中のDNA配列について、bias dataを計算し、そのbias dataと、"DNA配列中の各部分"のbias dataと比較し、bias differenceを算出します。またこの際、比較に用いるbiasデータとして、"ファイル中の配列のcoreのbias data"を選択すると、core bias dataと比べたときのdifferenceが計算されます。ここでcore bias dataは、指定したDNA配列を指定のwindow size、step sizeで各部分につてbias difference をもとめ、その値が小さかった上位50%について計算したbias dataです。つまりDNA配列全体の傾向と異なる配列組成を持つ側半分を捨て、残った半分で再計算されたbias dataに対するdifferenceを得ることができます。また、任意のbias dataを指定しそれに対する差を計算することも出来ます。また、解析するDNA配列の入力方法として"テキストフィールド"を指定すると、配列入力欄に入力したDNAそれぞれについて、指定のbias dataとのbias differenceを計算できます。
バージョン1.21から1.25への変更点は以下の通りです。
- 設定画面(コマンド+Bで開く画面)で、相同性(%)の分布を示すヒストグラムを見ることが出来るようにしました。階級幅は1%です。ヒストグラムをコントロールキーを押しながらクリックすると数値データにアクセスできまたPDF形式のヒストグラムを得ることができます。
- SNPを表示する機能を付けました<実行例>。MUMmerによって検出された点変異の位置を図示します。変異のうち、transitionを白で、transversionを緑で、indelを青で示します。SNPはMUMmerによって検出しますので、MUMmerをインストールし、MUMmerプログラムの入ったフォルダへのパスを指定する必要があります。
- カテネーションモードに変更を加えました。 今までカテネーションした配列とそれを構成する配列の長さのリストをユーザーが入力しなくてはなりませんでしたが、ファイルを指定することで自動的に、1)塩基配列の連結、2)それぞれの配列の長さのリスト、を作成する機能を追加しました。x軸、y軸それぞれについてadd filesボタンを押して、ゲノム配列ファイル(GenBank形式、FASTA形式、テキスト形式)を指定してください。複数のレプリコンより構成されるゲノムの場合は、複数のファイルを選択してください。複数のゲノムを指定するには、この操作を繰り返してください。かならずしも望む順序ではファイルパスが並びませんので(version 1.282より大きいレプリコン順に並ぶように変更)、必要に応じてファイルパスをテキストエディットあるいは表計算シートなどで編集してください(ゲノムとゲノムは「改行」でレプリコンとレプリコンは、「
カンマ」で区切られている必要があります(version 1.270より「タブ」に変更)。ファイルパスを指定できたら、"creat and set concatenated sequence"ボタンを押してください。注釈付のファイルの場合は、結果に反映されます。
それ以前のバージョンからの変更点は以下の通りです。
- メニューや表示されるヘルプが日本語になりました(言語環境が日本語のとき)。
- 画面中に表示されている範囲のアノテーションデータを一度に見られるようにしました。エクセルシートに貼り付けるための形式で出力できます。GenBankファイルのデータをエクセルシートで閲覧しやすい形式に変更するのにも使えます。塩基配列データも得られます。「解析」メニューから「現在の範囲のアノテーションを見る」項目の中のサブ項目を選んでください。
- MUMmerが利用できるようになりました(MUMmerをインストールをする必要があります)。MUMmerの結果が2次元に描画されます。アラインメントも表示できます。
- MAFFTが利用できるようになりました(MAFFTをインストールをする必要があります)。
- 表示画像をシフトを押しながらドラッグすることで解析範囲を変更できるようになりました。
- 進行状況を表すプログレスインディケーターを画面の右下に付けました。
- GUIには基本的にツールチップを付けました(ポインタを重ねるとヘルプが表示されます)。
- メイン画面にあった実行結果をPDFファイルへ書き出すためのボタンを削除し、Imageメニューから実行するようにしました。
- 付属機能として、遺伝子お絵かき機能を付けました。アクセサリーメニューから起動してください。この機能ではメイン画面で読み込んだ注釈データに加えて4種類のデータを追加入力することができます。1:遺伝子情報、2:鉛直線、3;水平線、4:文字列。それぞれ指定した位置に書くことができます。PDF形式で書き出せるので各種の描画をする際の下絵を作成する機能として活用できますフィードバックをお待ちしています。
- 付属機能として、"RecordMatcher"を付けました。アクセサリーメニューから起動してください。"key"と"value"の1対1の対応関係が複数与えられているときに、keyを指定するとvalueを得ることが出来ます。keyは複数を同時に指定することができます。例えばCOG番号それぞれにはProdduct名が1対1で関係づけられていますが、手元にCOG番号が1000個ありそれらのProduct名を知りたいときに使います。フィードバックをお待ちしています。
- 付属機能として、multi-FASTAモードを付けました。アクセサリーメニューから起動してください。1つのリファレンス配列といくつかのコンティグとを比較するために作成した機能です。y軸に読み込んだ配列(メイン画面で指定)とmulti FASTA形式のファイルに含まれる複数の配列(専用の画面で指定)を比較できます。後者では注釈付のファイルは読み込めません。描画されたイメージをコントロールキーを押しながらクリックしてみてください。メイン画面に持って行ってより詳細に解析することなどが可能です。フィードバックをお待ちしています。
- 遺伝子マークの色とタイトルを編集できるようにしました。コマンドキーを押しながらクリックすると編集用のウィンドウが開きます。
以下のステップに従うことでGenomeMatcherの基本的な使い方がご理解いただけると思います。
練習用に2つの塩基配列データを使います。以下の2つの配列をダウンロードしてください。これらのデータはDDBJが提供するGenome Information Broker (http://gib.genes.nig.ac.jp/)から2008年3月31日にダウンロードしたデータです(なおこのページからは多種のゲノム配列が簡単にダウンロードできます。GenoemMatcherを走らせるにはうってつけです)。
ステップその一: 塩基配列と注釈情報を読み込む
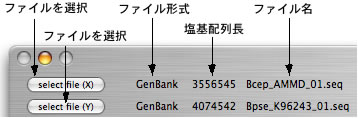
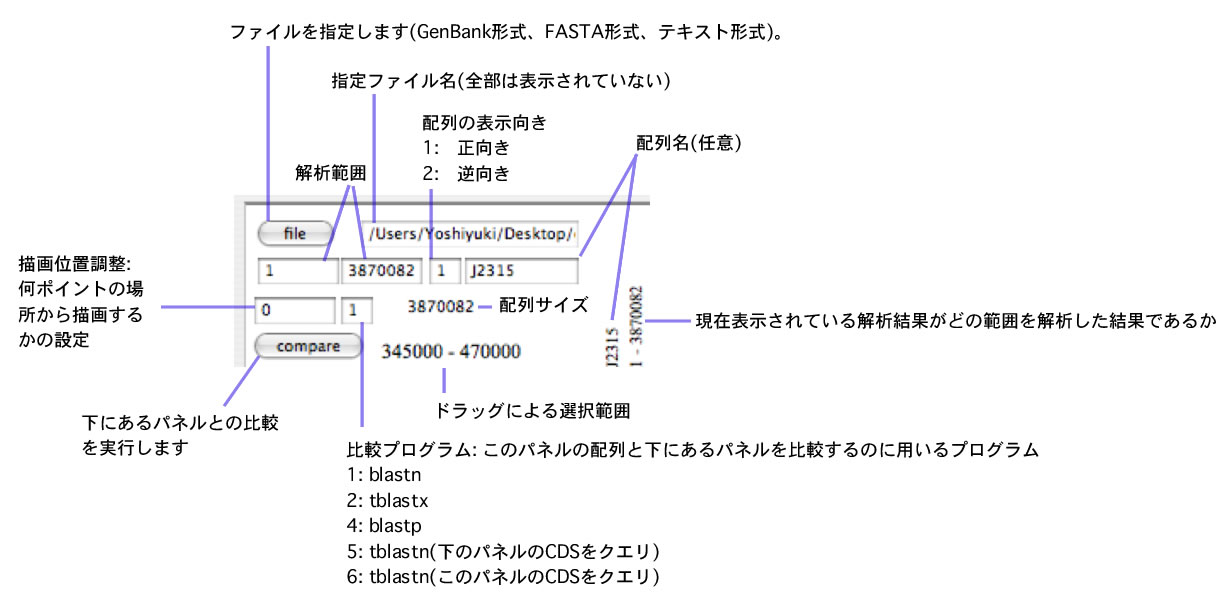
ファイルから塩基配列データを読み込みます。 画面左上の2つのボタン "Select File(X)"と"Select File (Y)"をクリックしてそれぞれ入力してください。塩基配列が読み込まれると、入力ファイルの形式、塩基配列長、 ファイル名、がそれぞれ表示されます。入力用ボタンそれぞれについて、前回使用したフォルダーが記憶されています(初期値はユーザーフォルダ)。
ステップその二: bl2seqを実行して比較図を得る<実行例>
2つの塩基配列を読み込んだ後に"Compare" とかかれたボタンをクリックするとbl2seqが実行され、しばらく後に比較イメージが表示されます。イメージの左下が原点です。出力までの時間はパソコンのスペックによって異なります。マニュアルのトップに機種別の解析時間の目安があります。出力された線の色は、blastnによる相同性を反映しています。右側にあるカラースケールが色と相同性の関係を表しています。このスケールはコマンド+Bで開くウィンドウ中で編集できます。
![]() カラースケール。90度回転させています。PDF形式で出力すれば編集することができます。
カラースケール。90度回転させています。PDF形式で出力すれば編集することができます。
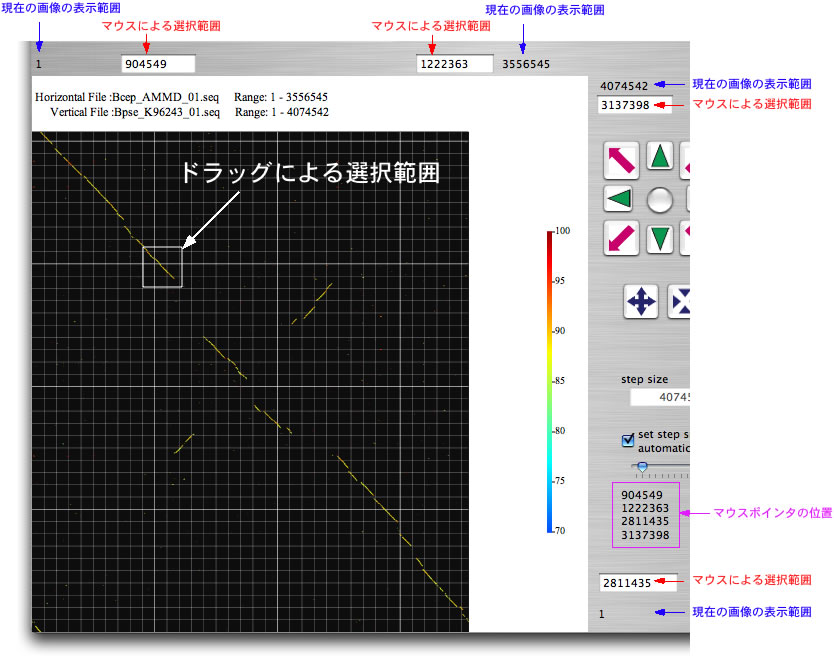
ステップその三: さらに解析したい領域を選択する
得られた比較イメージの細部をさらに解析するために、解析したい部分をドラッグします。ドラッグ中は、マウスポインターの位置が画面右下に表示されています。 新しい解析範囲は4つのテキストボックスに表示されます。新しい解析範囲はキーボードから手入力することも出来ます。新しい範囲を指定したら、再びbl2seqを使って解析して見てください。また8方向の矢印よりなるナビゲーションボタンをクリックすると解析範囲が平行移動した上でbl2seqによる解析が実行されます。平行移動する距離は、step size テキストボックスで指定されています。また、シフトキーを押しながら画像をドラッグすると、解析範囲がドラッグした方向にドラッグした距離に応じて移動した上でbl2seqによる解析が実行されます。
注釈情報が読み込まれておりまた、現在の表示幅(bp)が設定値(コマンド+Bで変更できます)以下の場合は、注釈情報を意味する矢印あるいは長方形が比較イメージ周辺に現れます。これらをクリックすると注釈情報が表示されます。また「解析」メニューの中の「現在の範囲のアノテーションを見る」項目の中にあるサブメニューを選択すると、現在表示されている範囲のアノテーションデータをまとめて参照できます。データはx軸y軸の配列それぞれについて2通りの形式で得ることができます。一つは、GenBank/DDBJ形式で、もう一つは表計算ソフトで編集するのに適した形式です。なお後者は、GenomeMatcherが受け付けるアノテーション形式とほとんど同形式となっています(11列あるデータのうち最後の2列が余分です)。表計算シートにペーストしてみて下さい。
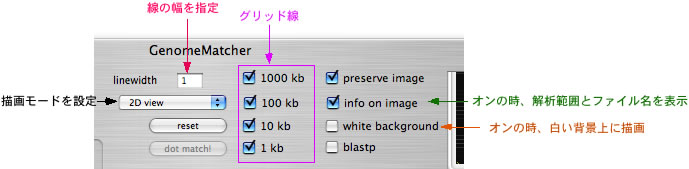
ステップその四: 描画イメージの見た目を変更する
描画イメージに関わる設定を変更することでイメージの見た目を変更することができます。各種グリッド線の有無、背景を白にするかどうか、解析範囲とファイル名を画像に記録するか、といった点を変更できます。また通常の描画モードは"2D view"ですが、parallel viewあるいはparallel view (flip)を選択することで2つの配列を平行に配置した描画イメージを得ることができます。parallel viewあるいはparallel view (flip)を用いた場合、下側の配列がx軸上にあった配列、上側の配列がy軸上にあった配列です。
ステップその五: 描画イメージを保存する
保存には2通りの形式があります。一つは既に描画されているイメージを、TIFF形式、PNG形式、JPEG形式のいずれかで書き出す形式です。この場合、「イメージ」メニューから「メインウィンドウのイメージを保存」項目を選択(あるいはコマンド+S)してください。この時、保存されるイメージはベクトル形式ではありません。もう一つは、再解析を行って、得られる描画イメージをPDF形式で描き出す形式です。この場合、「イメージ」メニューから「解析して結果をPDFヘ」を選択してください。こちらで保存した場合は、保存されるイメージはベクトル形式なので、お絵かきソフトで自由に編集が可能です。後者の場合、現在表示されている画像ではなく新たに解析して得られる画像が、PDFファイルに保存されることにご注意下さい。
ステップその六: アラインメントを得る<実行例>
選択範囲の配列について各種アルゴリズムを用いたアラインメントの取得が可能です。アラインメントに用いるプログラムを選択すると自動的に解析が開始します。mummerとbl2seqについては2つの配列の向きによらず解析ができますが、MAFFTとClustalWの場合は2つの配列の関係によってmafft-reverseあるいはClustalW-reverseを選択してください。MAFFTとClustalWには制限値があります。これを変更するにはコマンド+Bで設定画面を開いてください。

ステップその七: DotMatchを使う<実行例1><実行例2>
範囲を選択後、dot matchボタンを押してください。
DotMatchでは2つの閾値が設定できます。一つめの閾値より長いマッチは黒で、二つめの閾値より長いマッチは赤で示されます。
DotMatchでは比較範囲は常に正方形となります。
コマンド+Zでやり直しができます。
解析範囲が100bp以下になると塩基配列が、比較図に沿って表示されます。
- シーケンスの入力
- アノテーションの入力
- 文字列と色の設定
- カラースケールの編集
- パラレルビューモード
- アラインメント
- X モード と Y モード
- シンテニー解析
- 連結配列の比較
- Dot match解析
- 矢印の色の変更方法
- 外部配列でサーチ
- blastp総当たり比較
- カラーグラム
- 保存の仕方
シーケンスの入力(受け付けるシーケンスフォーマット)
GenomeMatcherが受け付けるファイル形式は
i) DDBJ / Genbank形式 (ただし塩基配列の直前に"ORIGIN"と一度だけかかれているもの)、
ii)FASTA 形式(multi-FASTA形式不可)、
iii)テキスト形式、
です。
独自アノテーションデータの入力方法
ユーザは独自の注釈情報を読み込ませることが出来ます(コマンド + Mで専用のウィンドウが開きます)。すでに注釈情報が入力されている場合は、既存のデータにデータが追加されます。既存のデータを消去するには、「Remove all entries」ボタンを押してください。
受け付ける注釈情報は以下の7つの値がタブで区切られたものを1セットとして、複数のセットが改行キーで区切られたものです( 難しく考えるまえに下にある表計算シートを見てください)。7つの値はそれぞれある一つの情報単位の i)開始位置、 ii)終了位置、 iii) 向き +1(順向き)または-1 (逆向き)または0 (向き情報なし)、 iv) 注釈情報、v) DDBJ / GenBankの形式に準拠するフィーチャーキー、vi)遺伝子ボタンにつけるタイトル、vii)色を表す文字列、です. 終了位置は、開始位置よりも大きくなるようにして下さい。最初の2つの値は必須ですが、残りの5つの値は指定しなくても構いません。vについての補足:それぞれのfeature_keyについて、表示、非表示の設定が可能となっています。つまり表示する設定になっていないfeature_keyを指定してしまうとその行のデータは表示されなくなってしまいます。表示、非表示の設定についてはコマンド+Bで開く設定画面を見てください。viiについての補足:「色」と「文字列」の関係は、コマンド+Kで開くウィンドウ中で編集してください。この値が空白の場合はvの値が代用されます。vの値が空白の場合はデフォルトの色が使用されます。デフォルトの色はこのウィンドウ中の右上にあるカラーセルで定義されています。サンプルとして注釈情報を編集したエクセルシートをダウンロードできます。これのもととなる注釈ファイルはこのリンク先からダウンロードできます。
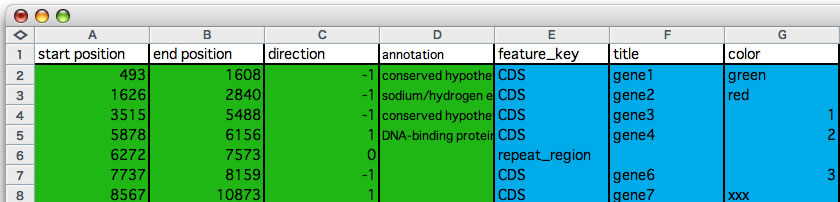
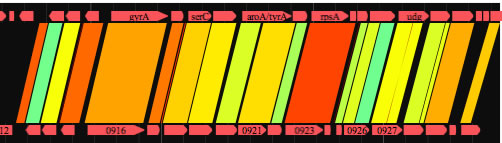
着色部分をコピーして利用する。AB列は必須。タイトル行を含めないように注意。
コマンド+Kで色と文字列の関係を定義するための設定ウィンドウが開きます。このテーブル中、すべての文字列はユニークである必要があります(ユニークでない文字は入力できません)。右上にあるカラーセルはデフォルトの色を表しています。このテーブルで定義されていない文字列が色を指定する文字列として指定したときにこのデフォルトの色が使用されます。
カラースケールの色調は3つの数値で決定されています。6つの値がそれぞれ何を指定しているかについてはこの図を見てください。
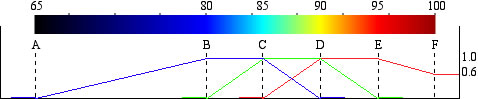
カラースケールを編集する上でのヒント. ここでは例として6つの値(65、 80、 85、 90、 95、および 100 )に依存して作成されたカラースケールを示している.この図に示したようにRed Green Blueが配合されている.縦軸は各色の強度(1.0が最大値)。
GenomeMatcherでは比較結果を2通りの方法で表示させることができます。一つは2つの配列を縦横に垂直方向に配置して、その配列間の相同性を示す方法です。もう一方は2つの配列を平行に配置して、相同性を示す方法です。後者の表示をさせるには、 "parallel view"または"parallel view (flip)" を、プルダウンメニューより選びます。下の配列がX軸方向に置いていた配列、上の配列がY軸方向に置いていた配列です。"parallel view" モードでは、どちらの配列も左側から置かれます。"parallel view (flip)"モードでは上の配列について右側から置かれます。parallel viewモードの場合、つぎの解析範囲を設定するには解析したい部位を2つの配列それぞれについてドラッグすることで指定してください。



パラレルビューによる表示例。一番下は連結モードで解析した例。青線がレプリコンの境界を示している。
アラインメント<実行例>
ヌクレオチドのアラインメントを得ることができます。このポップアップボタンから用いたいプログラムを選択してください。プログラム名がグレイアウトしている場合は利用できません。これは現在の解析範囲が制限値を超えているためです。制限値は時間のかかる解析をユーザーが始めてしまわないようにするために設定されています。制限値はコマンド+Bで開く設定ウィンドウで変更できます。アラインメントについてはこちらも参照してください。
Xモードおよび Y モードと名付けられた比較モードを利用すると離れた2つの領域を近づけて眺めることが出来ます。これらのモードを呼び出したのち、ドラッグすると正方形の半分の大きさの長方形が現れます。ドラッグを止めた後再度比較イメージの上でクリックすると先の長方形と同じ形の長方形が現れます。これをドラッグして適当な場所に置きます。別のウィンドウが開き、指定した2つの領域の比較結果がそれぞれ表示されます。
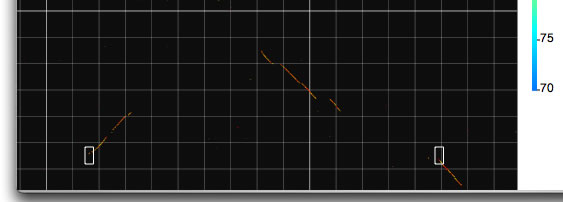
Xモードを使用して2つの領域を指定したところ。 2つの白い長方形が選択された領域
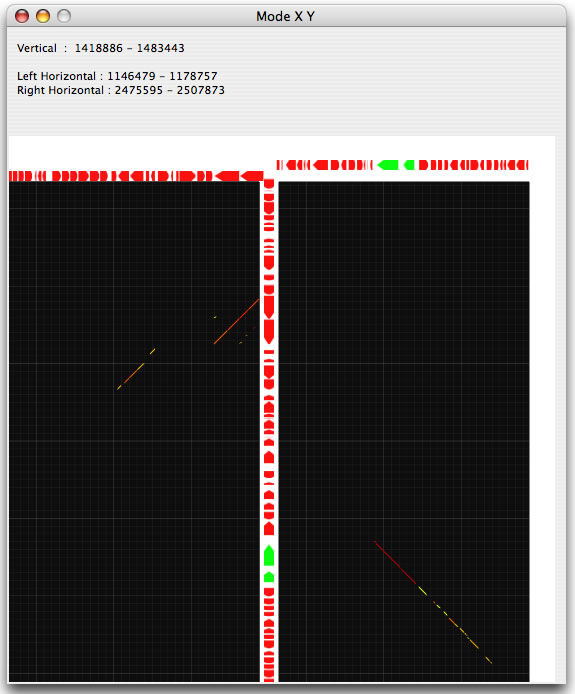
Xモードによる比較結果.
GenomeMatcherは、2つの配列中に複数のHSPが並んでいる領域があれば、その領域を連結して表示すると同時に、連結された領域の位置情報をテキスト形式で出力する機能を備えています。シンテニー解析のスイッチが有効になっていると、GenomeMatcherはまずテキストボックスで定められた感度に従って、近くにあるHSPを連結します.複数のHSPが連結されたものをcHSP(connected HSP)と呼びます。cHSPsは太い青い線で示されます。GenomeMatcherはさらに複数のcHSPsを、ある値( 固定値:20kb)をしきい値として連結します。連結されたcHSPをccHSP(connected cHSP)と呼びます。ccHSPsは紫色の折れ線で示されます。 また結果はテキスト形式でも出力されますが、テキスト形式では、ccHSPの開始座標および終了座標に始まり、ccHSPを構成するいくつかのcHSPの開始座標と終了座標が出力されます。ユーザはこの解析結果が適正であるかどうかを、出力された図を眺めることで検討することができます。
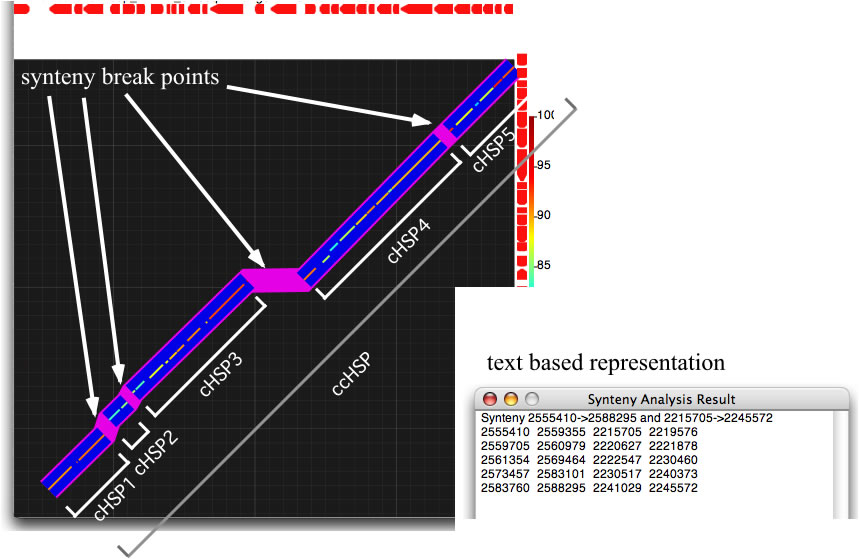
GenomeMatcherによるシンテニー解析。 シンテニーがとぎれる部分は紫色の帯として表示される.テキスト形式の出力については下図を参照のこと。
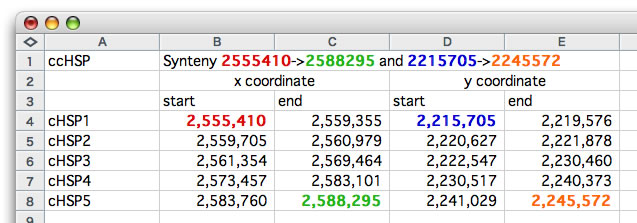
テキスト形式の結果の見方.同じ値には同じ色を付けて示した。
連結して作成した配列の比較<実行例>
<version 1.25から複数の配列を自動で連結する新しい機能がついています。>
ある種のバクテリアは複数のレプリコンを保持しています。マルチレプリコンよりなる2つのゲノム配列を簡便に比較する手段として、比較に先立ってそれぞれの細菌の複数のレプリコンの配列を連結することが行われています。"catenation モード"では比較対象の連結塩基配列について、それを構成するレプリコンのサイズを指定することでレプリコンごとに比較することができます。また、比較によって描かれる図は、レプリコンごとに長方形で囲まれるため、レプリコンの境界を判別することが容易です。 画面の下に解析の進行状況が表示されます。時間がかかる解析を実行するときには参考になるでしょう。
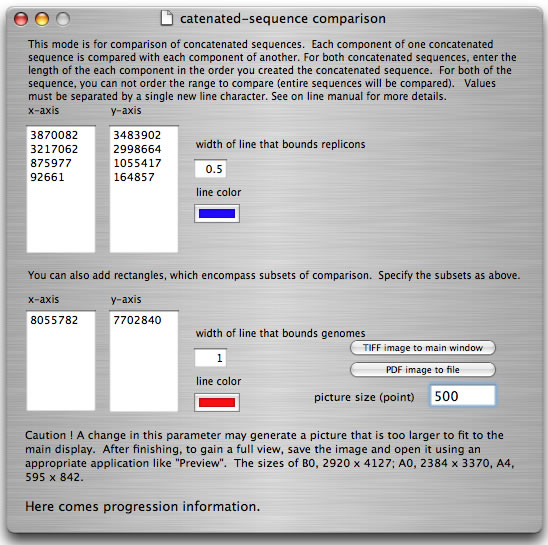
catenation モードの使い方。比較しようとしている2つの配列は両方とも4レプリコンより構成され、そのうち一方のレプリコンサイズは3,870,082 bp、 3,217,062 bp、 875,977 bp、 92,661 bp (total 8,055,872 bp)である。またもう一方のサイズは 3,483,902 bp、 2,998,664 bp、 1,055,417 bp、 164,857 bp (total 7,702,840 bp)である。メインウィンドウで2つの配列 (それぞれ8,055,782 bpおよび7,702,840 bp)をロードした後にこのセッティングウィンドウを開きここに示したように値を入力する。ボタンをクリックすることで解析が開始する(出力はPDFファイル、あるいはメイン画面へと出力される)。

catenationモードによる比較結果。レプリコンの境界の線の色(青)はセッティングウィンドウで指定した色である。
catenationモードは連結した複数のゲノムの比較にも用いることができます。この場合、連結したレプリコンのサイズに加えて、それぞれのゲノムサイズを指定します。ここでは3つの細菌ゲノム(それぞれマルチレプリコンよりなる)を連結したものを、それ自身と比較しようとしている場合を例を示します。連結した配列を連結した順に以下に示します.Burkholderia cenocepacia J2315 (4レプリコン、3,870,082 bp、 3,217,062 bp、 875,977 bp、 92,661 bp; 全部で8,055,782 bp)、B. pseudomallei K96243 (2レプリコン、 4,074,542 bp、 3,173,005 bp; 全部で7,247,547 bp)、B. xenovorans LB400 (3レプリコン、4,895,836 bp、 3,363,523 bp、 1,471,779 bp; 全部で9,731,138 bp)。
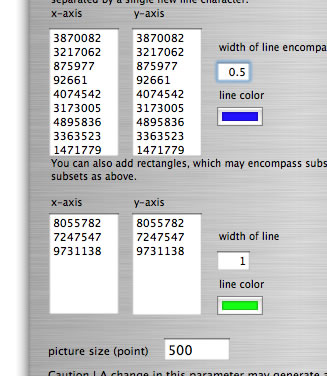
連結したゲノム配列をそれ自身と比較するときのセッティング例.
ピクチャーサイズを500ポイントから変更することでより大きい比較イメージを描画することが出来ます。例えばA0サイズにするには幅を2300ポイントにします。ただし、大きい比較イメージを作成した場合、Main画面に収まらなくなってしまいます。この場合、描画した絵はtiff/png/jpeg形式でファイルに書き出して(コマンド+S)適当な描画ソフトウエアを使って眺めるか、最初から結果をPDF形式として書き出すようにして下さい。
メインウィンドウからDotMatchを開始するには選択されている範囲が10 kb以下でなくてはなりません(このサイズはコマンド+Bで開くウィンドウで編集が可能です)。ただしdot matchを開始した後はこの制限値を超える範囲の解析が可能です。dot matchによる解析は以下に述べる特色により 比較結果を簡単に得ることができます。同じ2つの配列を比較することで、1)挿入配列(IS)の末端形状の解析、2)転写ターミネーターの同定、3)ori部位の同定、4)繰り返し配列(転写因子の結合部位など)の同定、5)integronの同定、6)CRISPRの同定、を行うことができます。
1) 2つのワードサイズが設定可能です。1つ目のワードサイズを利用した結果は黒線で示され、2つ目のワードサイズの結果は赤で示されます。このため特に長い範囲が比較されている場合に、意味のある相同領域を見分けることが容易です。
2) 一度に両方向の比較が可能で結果は同一のウィンドウ上に表示されます。どの向きを比較するかはスイッチによって設定を変えることができます。
3) 比較に用いられた配列がテキストボックスに表示されます。また特に 鉛直方向に配置された配列についてはその相補配列が同時に表示されます。このため実際に比較に用いられた範囲の塩基配列データを速やかに手にすることができます。
4) 比較に用いている配列が100 bp以下になると、塩基を表すA、 C、 G、 Tの文字がグリッド線にそって表示されます<実行例>。
5) 画面をドラッグするとその範囲が解析されます。
6) ClustalWを実行することが出来ます。
7) 現在開いている解析ウィンドウと同一のウィンドウを開くことができます。2つのウィンドウはそれぞれ別々に解析することが可能なため比較的離れて存在している短い配列の繰り返しを探すことが容易です。このような繰り返しは、挿入配列やゲノミックアイランドの周囲に観察されます。
8) 解析範囲は8方向のボタンに加え、拡大ボタン、縮小ボタンをクリックすることで簡単に変更できます。また解析範囲は開始座標および比較解析長を入力することでも設定できます。
9) アンドゥ(動作の取り消し)ができます(コマンド+ Z).何度でも出来ます。
10) スイッチが有効になっていると、注釈情報を表す矢印が表示されます。矢印をクリックすると注釈情報がテキスト形式で表示されます。
矢印の色の変更方法<実行例>
GenomeMatcherでは「文字列」で色を指定します。文字列とは例えば'Red'とか'Green'等ですが、'1'や'A'など任意のものが利用できます。各文字列が実際にどんな色を意味しているかは、コマンド+Kで開くウィンドウ中で設定する必要があります。すでにいくつかの関係が定義されていますが、add newボタンを押すことでさらなる関係を定義することができます。このウィンドウの中で各文字列はユニークとなるようになっており、ユニークでない文字列は入力することができません。この「文字列」と「色」の関係を変更することで描画される遺伝子を表す色を変更することができます。
外部配列を用いたサーチ<blastp実行例><モチーフサーチ実行例>
読み込んだ配列に対して、任意の配列を問い合わせ配列として相同性検索を行うことができます(解析メニュー→位置を探す)。問い合わせ配列として用いることができるのはDNA配列またはアミノ酸配列です。相同性検索の種類はプルダウンメニューより選択してください。利用できるのはblastn、blastp(内部にアミノ酸配列が生成されているときのみ)、tblastnです。これらのBLAST検索に加えて、検索配列と完全一致する部位を全件表示する「simple find」機能と、検索配列と似た部位を全件表示する「motif find」機能を利用することができます。「motif find」では許容するミスマッチの数を右上にあるテキストボックスで指定します。注意:「motif find」は極めて時間のかかる機能です。利用する場合は短い対象配列に対して短い検索配列をミスマッチを余り許容せずに試行するなどして必要となる時間を把握した上で利用してください。
blastpによる総当たり比較<実行例>
blastpとかかれたチェックボックスをオンにしておくと画面内に表示されているCDS間で総当たりでblastpが実行されます(ただし総当たりの回数が一定数(初期値900)を超えるときはかかりません(コマンド+Bで開くウィンドウで変更できます)。ヒットがあれば2つのCDSの交差する部位に四角形が表示されます。四角形をクリックするとblastpによるアライメント結果を見ることができます。blastpのパラメーターはコマンド+Bで開くウィンドウ中で設定できます(MacOS10.4では四角形をクリックするとblastnの描画結果が変にうつりこんで表示されます。10.5では正常です)。また、blastpの結果がテキスト形式で得られます。そのうち片方は、表計算シートにペーストするのに適した形式です。
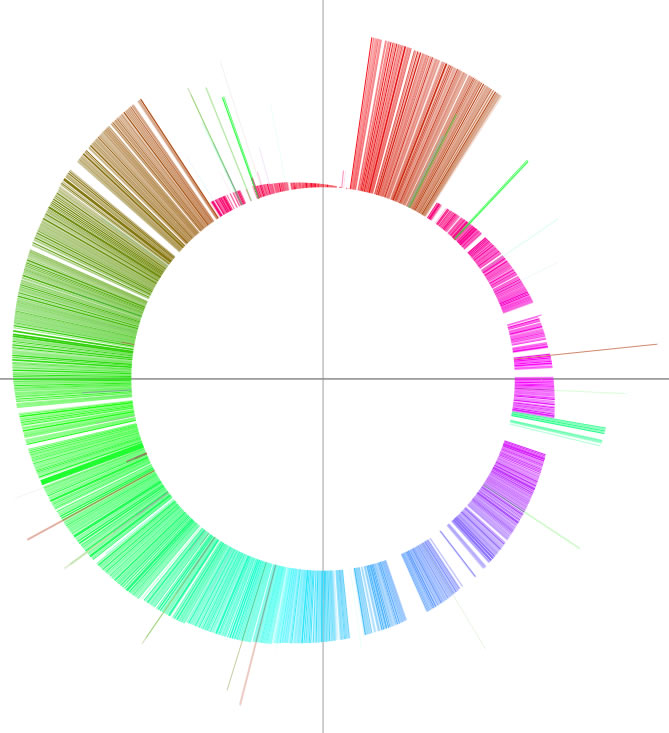
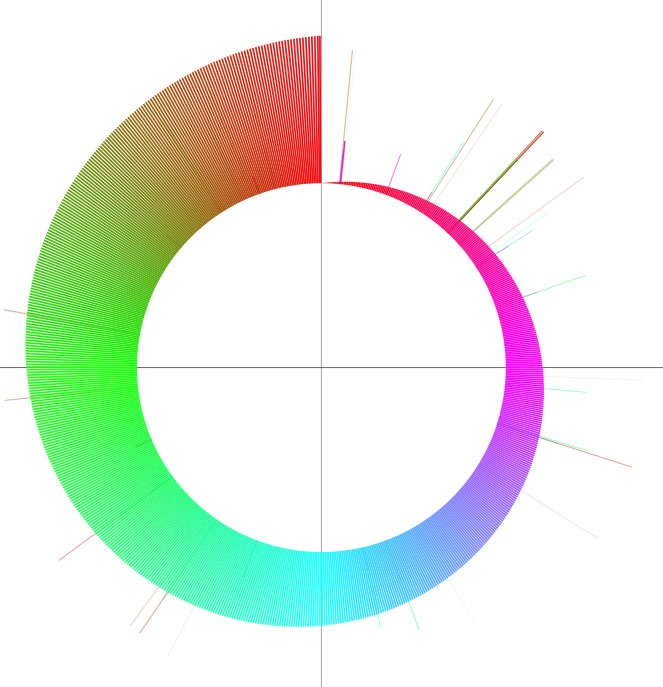
カラーグラム<実行例>
2つのゲノムの形の違いを表現するための機能です。2つの配列の全長が解析範囲になっている状態でcolorgramとかかれたボタンをクリックしてください。X軸上に読み込んでいる配列(ここではX軸上に配置されているわけではないのでA配列と呼びます)の各部分が、Y軸上に読み込んでいる配列(同じくB配列と呼びます)のどの部位に位置しているかが表示されます。A配列は0時から時計回りに配置されています。カラフルな線がたくさん現れると思いますが、線の「高さ」と「色」は両方ともB配列のどの位置に相同配列が存在するか、を表しています。一度の逆位によって形状の違いの説明がつく2つのゲノムの比較を例として示します(左)。またある配列をそれ自身と比較した結果も併せて示します(右)。描画ウィンドウ上のテキストボックス中の値を変えることで絵の見た目を調整することができます。また右側にある大きめのテキストボックスに特定の形式に従ってデータを入力することで、任意の位置に直線と文字列を追加することができます。この形式は、「線を引く位置(bp)」「線の高さ(単位はポイント)」「表示したい文字列(任意)」の3つの値をタブ文字で区切ったものです(表計算ソフトで編集すればいいのですが、数が少ないときは直接入力すると良いでしょう。テキストボックス中でタブ文字を入力するときはオプションキーを押しながら「tab」キーを押します。改行は同様にオプションキーを押しながらリターンキーを押します)。
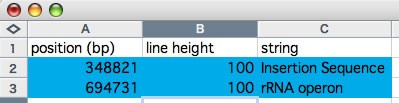
カラーグラムに追加するデータの編集例。青い部分をコピーして使う。
「イメージ」メニュー中の「解析して結果をPDFへ」を選択すると、実行結果がPDF形式のファイルとして書き出されます。また、ドットマッチウィンドウにある"PDF screen shot"ボタン、連結モードの設定ウィンドウにある"PDF image to file"ボタンをクリックしても同様です。X Yモードの結果をPDFファイルとして書き出すには「イメージ」メニューから"X Y モードのイメージを保存"を選んでください。また、メインウィンドウの画像およびドットマッチによる画像はTIFF形式でも書き出すことが出来ます。ファイルメニューから対応するコマンドを選択してください。
メインウィンドウの右側にあるイメージカラムを保存することができます。保存したイメージカラムは読み込むことで再び解析に用いることが出来ます。将来的には改良する予定ですが、イメージカラムに画像がたくさんあると、とても大きなファイルになってしまいます。不要なイメージを削除した後に最低限のイメージを保存とするようにしてください (イメージカラム中の画像はコマンド+Dで消すことが出来ます).
GenomeMatcherには便利な機能がいくつか付属でついています。便利機能は今後拡充する予定です。ゲノム解析に限らずご活用ください。なおこのマニュアルは機能の付加と平行して作成しているため、実際に公開している最新バージョンに該当する機能が付加されていない場合があります。あらかじめご了承ください。またこれらの付属機能についてもユーザーの皆様の声を反映させて使いやすくしていきたいと思っていますので、フィードバックをいただければ幸いです。
この機能の概略
この付属機能は遺伝子などの絵を描くのに特化した機能で、DNA配列の比較とは関係がありません。メイン画面で読み込んでいる注釈付配列のいずれか一方のデータ(sourceタブ中で選択)に加え、1)遺伝子を表す5角形あるいは長方形、2)鉛直方向の線、3)水平方向の線、4)文字列、についてデータを指定することで描画することができます。これらのデータは表計算シート上でデータを編集し、コピーペーストすることを想定しています。またこれらのデータのy軸方向の描画位置について、専用の「drwaing height」欄で指定/変更することができ、グラフィックを見ながら描画位置を調整することができます。描画した絵はお絵かきソフトで編集可能なPDF形式で出力することができます。
データ形式
データはそれぞれの種類について3セットまで指定できます。データはそれぞれ決まった形式に従っている必要があります。データ形式についてはGeneDrawerの画面中に概略が記してあります。1)についてはGenomeMatcherのアノテーションの入力方法と一緒です。2)は描画位置(bp)、描く線の長さ(ポイント)、線の太さ、線の色を指定します。線の長さはdrwaing heightで指定した高さ(ポイント)を下端として描かれます。3)は水平線の開始位置(bp)、終了位置(bp)、drwaing heightで指定した下端位置からの相対的高さ(ポイント)、線の太さ(ポイント)、線の色を指定します。4)はすこし特殊で、最初の2つのデータで位置を指定しますが実際に描画されるのはこれらの中間点です。3つめのデータとして描画する文字列、4つめのデータでフォントサイズ、5つめのデータで色を指定します。いずれの場合もデータ中の値はタブ文字で区切られている必要があります。また複数のデータを改行文字区切りで複数指定することができます。要するに表計算シートで編集したデータセットを一度に入力することができます。
データの入力方法
テキストボックスにデータを入力したら、addボタンを押してください。テキストボックスの背景が青色になります。青色はそのセルに入力したデータが有効になっていることを示しています。addボタンを押す度にテキストボックス内のデータが追加されます。一度addボタンで入力したデータはremoveボタンを押さない限り消されません。removeボタンを押すとそれまでに入力したデータはすべて破棄され、背景色が白になります。
描画方法
左上の2つのテキストボックスで描画する位置範囲を、またそのとなりのテキストボックスで描画比率を指定してください。drawボタンを押すと描画されます。またsave in PDFボタンを押すとPDF形式で保存することができます。拡大したい部位をドラッグすると拡大できます。拡大しすぎたらzoom outボタンでズームアウトしてください。GenomeMatcherと同様、遺伝子シンボルの色とタイトルはコマンドキーを押しながらクリックすると変更することができます。
データの保存
一度入力した4種のデータは保存することができます。再読込もできます。
この機能の概要
1つの配列を複数の配列と比較することができる機能です。塩基配列の解読が終わっている配列をリファレンスとして、塩基配列を決定途中のcontigs(あるいはsingle reads)とを比べることを目的に作成しました。1対多の比較イメージの中から任意のものを選択してGenomeMatcherのメイン画面に移行して解析を続けることもできます。
配列の指定
GenomeMatcherのメイン画面で、y軸にリファレンスとなる配列を読み込んでください。もう一方の配列はmulti-FASTAモード画面の中でファイルを指定してください。この配列はmulti FASTA形式でなくてはなりません(注釈情報は受け付けません)。
操作
描かれたイメージにはコンテクストメニューが設定されています。コントロールキーを押しながらイメージをクリックしてください。「reverse if」ボタンを押すと、相補鎖側に相同性が見られる場合に、配列を相補鎖に変換します。「line up」ボタンを押すと、配列が自動で並び変わります。これらの精度は余り高くないのでマニュアルで修正してください。イメージは選択して、矢印キーで動かすことができます。複数のイメージはコマンドキーを押しながら、あるいはシフトキーを押しながら選択してください。
この機能の概要
複雑で大きなタスクはより小さい単純なタスクの組み合わせに分割することを考えると解決することが多いと言えますが、単純なタスクでありながら普通に(コードを書かずに)パソコンを使っていてはなかなか解決できないタスクがあります。Record Matcherはそのような"単純なタスク"の1つを実行するための機能です。
Record Matcherを使用すると、1対1の対応関係が与えられているときに、その一方を指定してもう一方を取り出すことができます。例えば、犬→ほ乳類、亀→は虫類、カエル→両生類という関係が与えられている時、その関係を左側のテキストボックスに入力します(タブ区切り、改行キー区切り)。真ん中のテキストボックスで、カエル、カエル、犬、を指定すると、両生類、両生類、ほ乳類が右側のテキストボックスに表示されます。多数の1対1の関連が与えられており、また大量に調べたいデータがあるときに便利です。例えばCOG番号とプロダクトの関連を左端のテキストボックスで指定し、真ん中のテキストボックスで調べたいCOGを入力します。実行すると右側のテキストボックスにそのCOGのプロダクト名が表示されます。
データ形式
左側のテキストボックスに指定するデータはkeyとvalueのペアです。複数の関係を同時に指定できます。それぞれのペアは改行キーで区切られている必要があり、keyとvalueはタブ文字で区切られている必要があります。要するにこれらの関係は表計算シートで編集してコピーペーストすることが前提になっています。各行で最初に登場するタブ文字より左側にある文字列がkey、右側にある文字列がvalueと解釈されますので、valueとする文字列中にはタブ文字が含まれていても構いません。つまり、表計算シートで複数の列をvalueを意味する文字列として指定できます。同じkeyが複数ありそれらが異なるvalueと関連付けられている場合、最後に指定したkeyとvalueの関係が有効になります。
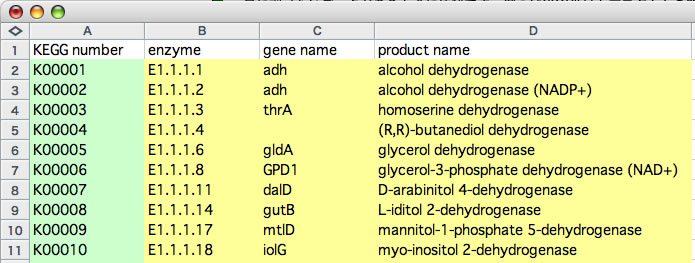
データ形式の例。着色部分をデータとして使う。緑色の部分がkeyとして、黄色の部分がvalueとして認識される。任意のkeyを指定すればvalueが出力される。
この機能の概要
複雑で大きなタスクはより小さい単純なタスクの組み合わせに分割することを考えると解決することが多いと言えますが、単純なタスクでありながら普通に(コードを書かずに)パソコンを使っていてはなかなか解決できないタスクがあります。Data Counterはそのような"単純なタスク"の1つを実行するための機能です。
2つのデータセットAとBがある場合に、1)Aのみに含まれる要素、2)共通で含まれる要素、3)Bのみに含まれる要素、を調べるための機能です。これらにについて定性的あるいは定量的に解析します。前者では、それぞれの要素の登場する回数は無視して解析されます。後者では、それぞれの要素について登場回数を考慮して解析されます。後者の機能を利用するとあるデータセットに含まれるそれぞれの要素についていくつあるかを知ることができます。1)ベン図の作成、2)更新されたデータの発見、3)処理前のデータと処理後のデータを比べることで、未処理データの抽出、等、使い方はいろいろです。
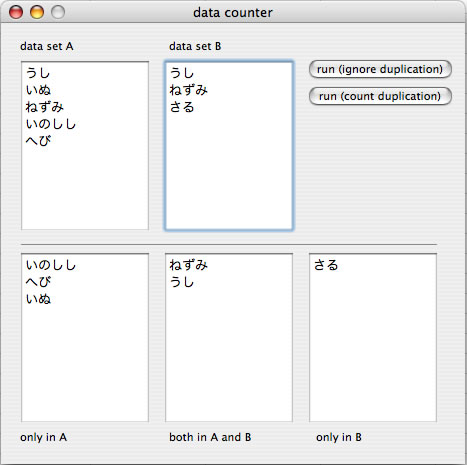
定性的な解析例
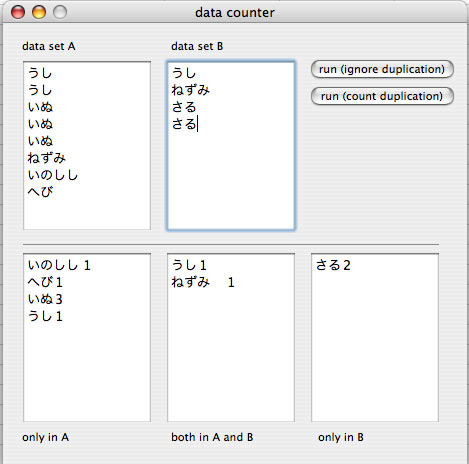
定量的な解析例。ここでは示していないが、データセットBを空欄にしておけば、データセットA中のそれぞれの要素の数を数えることができる。
この機能の概要 <画像をクリックすると大きい画像が表示されます>
複数の配列の指定範囲を各種blastプログラムで比較することができます。調べたい遺伝子の配列を用いてsearch and focus機能を使うと、読み込んであるそれぞれの配列の該当する箇所を指定範囲とすることができます。
概観
パネル制御部分

コンテクストメニュー
サーチ&フォーカス
アノテーション支援パネル
blastpの結果の見方
開始コドン位置決定機能
1:tblastxは非常に時間がかかります。特に長い範囲を比較すると時間を食います。100kb x 100kb 程度以内で比べると良いかと思われます。使ってみて様子を見てください。meshモードとくみあわせればゲノム全体をtblastxで比較したイメージを得ることが出来ます。
2:画面右側のイメージカラムを「ファイル」→「イメージカラムを保存」で保存することが出来ますが、比較イメージがたくさんカラムにあるとfileサイズがとても大きくなります。必要のないイメージを消去した上で保存するようにしてください。
3:Dot-Matchでは、X軸Y軸ともに終了位置を入力することができません。開始位置に、画面左上に示されているlengthの値が加算された値が終了位置に入力されるようになっています。
1:ACGT以外の文字が塩基配列データにある場合、トラブルが起きる可能性があります。
2:3の倍数にならない範囲をCDSとして登録しないでください。そのCDSは翻訳されなくなります。
3:注釈付きファイルの注釈の中に、配列の開始地点(1番の塩基)をまたぐ様に注釈が付されていると、エラーが起きます(矢印がきちんと表示されません)。注釈ファイルから該当する部分を取り除いてください。
4:長大な配列を、meshモードを使わないで解析しないでください。余りにも長い配列を一度に比較しようとすると目盛りが不足してクラッシュします。50 Mb x 50 Mbを一度に比較しようとしたらおそらくクラッシュします。
1:OS10.5ではイメージカラムの画像を矢印キーで選択できません。
2:OS10.3およびOS10.4では遺伝子マークをクリックした際に周辺に(その遺伝子マーク周辺の)画像が表示されることがあります。またblastpのヒットを表す四角形をクリックした際に周辺の画像が四角形内に表示されることがあります。
A:出力データは基本的に表計算シートにコピーペーストすることを前提に作っています。表計算シートにそのまま貼り付けられるように各データがタブ区切りになっています。そのため、出力されたデータそのものはきちんとしていないように見えます。
A:注釈付き配列情報を読み込んでいる場合、遺伝子名があれば遺伝子名、なければ、ローカスタグが表示されています。ユーザー独自のアノテーション情報を入力している場合は入力形式中6つめの値が表示されます。ユーザー独自データの入力形式についてはこちら。
A:十分な余裕がないときは文字列は表示されないようになっています。従ってユーザー独自のアノテーションを入力する際は短めの文字列を設定するようにしてください。
A:DDBJ/Genbank形式を入力した場合、表示されるのは初期状態では以下のフィアチャーキーに関連づけられた情報だけです:CDS、rRNA、tRNA、repeat_region、repeat_unit。これ以外の情報は表示されません。その他のフィアチャーキーに関連づけられた情報を表示したいときはコマンド+Bで開く設定ウィンドウ中で表示するフィアチャーキーを指定してください。また、描画範囲が広すぎる場合はシンボルが表示されないようになっています。初期値では200kb以下の時に表示されますが、この長さは変更することができます(コマンド+B)。またこの制限値は2つ設定できます。必要に応じて2つの制限値をうまく使ってください。
A:アノテーションデータの形式を見直してください。タイトル行(見出し)は含めないでください。
A:現在の設定が実行しようとする機能と相容れない場合、グレーアウトするようになっています。例えばシンテニー解析とX modeは相容れない機能ですので、シンテニー解析にチェックを入れるとX modeのボタンはグレーアウトします。気を付けて欲しいのはtblastxです。tblastxは解析時間を要するので解析範囲がある閾値を超えていると「Compare!」ボタンがグレーアウトします。閾値はコマンド+Bで開く画面中で変更できます(tblastxをmeshモードで使用するときはmeshサイズが閾値となります)。ファイルを2つ読み込む前はほとんどのボタンがグレーアウトしています。
A:パソコンに積んであるメモリに強く依存します。300Mbまで試したことがあります。長い配列をそのまま比較しようとするとbl2seqプログラムがクラッシュしてしまいますので、meshモードをご利用ください。また長い配列を解析すると全体が雲をかけたようになってしまいますのでe-valueをきつめに設定してください。
A:MUMmerとMAFFTはユーザーが自分でGenomeMatcherをお使いのMacにインストールした上で、パソコン上のどこにおいたのかをコマンド+Bで開くウィンドウ中で指定しなくてはなりません。
MUMmerは以下のURLから得ることができます。http://mummer.sourceforge.net/。ダウンロード後、解凍し、http://mummer.sourceforge.net/manual/の2.3. Compilation and installationにある記述に従ってください。
MUMmerをインストールするには、お使いのMacに、ソフトウエアの開発アプリケーションであるXcodeがインストールされている必要があります。XcodeのインストーラーはMac購入時に付属していたディスクに入っていると思います。まずこれをインストールしてください。
MUMmerは以下の手順でインストールできるかもしれません(ご自身の責任で行ってください)。
1:ダウンロード後、解凍。
2:解凍してできたフォルダ(ここでは名前は「MUMmer3.20」)を、"Macintosh HD"に入れる。
3: ターミナルを開く(ファインダーメニューで「移動」→「ユーティリティ」→「ターミナル」)。
4:コマンドラインに以下のアスタリスクに挟まれた部分をコピーしてペーストしてリターンを押す。****cd /MUMmer3.20/****
5:コマンドラインに以下のアスタリスクに挟まれた部分をコピーしてペーストしてリターンを押す。****make check****
6:コマンドラインに以下のアスタリスクに挟まれた部分をコピーしてペーストしてリターンを押す。****sudo make install****
7:パスワードを要求されるので入力する。
8 : 終了するまで待つ。
これでMUMmer3.20フォルダの中に必要な実行ファイルが作られます。GenomeMatcherに戻ってコマンド+Bで開くウィンドウ中で、フォルダの場所を指定してください。デフォルトでは、/MUMmer3.20/となっています。
A:MUMmerとMAFFTはユーザーが自分でGenomeMatcherをお使いのパソコンにインストールした上で、パソコン上のどこにおいたのかをコマンド+Bで開くウィンドウ上で指定しなくてはなりません。
MAFFTはhttp://align.bmr.kyushu-u.ac.jp/mafft/software/macosx.htmlよりダウンロードできます。ダウンロード後、インストレーションパッケージを利用してインストールを行ってください。デフォルトでは、mafftは/usr/local/bin/mafftにインストールされるようになっているようです。必要に応じてコマンド+Bで開くセッティングウィンドウでmafftのパソコン内での所在を指定してください。
できません。最新バージョンでは出来ます。イメージメニューから選択してください(コマンド+U)。TIFF、PNG、あるいはJPEGでも出力できます。
すみませんが簡単にはできません。平行描画によって得られる図を複数組み合わせてください。比較図は、長い方の辺が500ポイントです。 version 1.300より多対多の比較ができるようになりました。
新機能の要望、バグのレポートなどはこのアドレスにお願いします。e-mailの件名には「GenomeMatcher」の文字を入れてください。
また掲示板が設置してありますのでご利用下さい。
GenomeMatcherはアカデミックユーザーには無料で利用許諾いたします。無料ですが利用許諾契約書をよくお読みの上、許諾条項に同意の上ご利用下さい.非アカデミックユーザーはライセンスが必要です。一定期間の試用にも対応しています。お問い合わせ下さい。
GenomeMatcherを利用して得られた成果、あるいは作成した図を公表する際には、我々のGenomeMatcherについての論文を引用するとともにGenomeMatcherをダウンロードしたウエブサイトを明記してください.
GenoemMatcherの論文
GenomeMatcher: A graphical user interface for DNA sequence comparison.
BMC Bioinformatics 2008, 9:376 (16 September 2008)