DinucleotideBiasSignature
2塩基組成の偏りを計算する機能です。入力配列全体についてAAからTTまでの16通りの配列の出現頻度を調べ、16個の数値よりなるバイアスデータを算出します。これと、配列の各部分(例えば1kbの)のバイアスデータとの差異(バイアスディファレンス)を計算します。すなわち配列中の各部分の配列の組成が、配列全体の組成と比べてどれぐらい異なっているかについての値を与えます。外来領域の推定などに利用されています。
はじめに
DNA配列には生物に固有な偏りがあることが知られています。配列の偏りについてよく知られているのはGC含量ですが、連続2塩基の出現頻度についてもそのような偏りが存在します。「DinucleotideBiasSignature」を用いると2塩基出現頻度に関する解析を行うことができます。なおdinucleotide bias signatureについての詳細は原著論文を参照してください。
バイアスデータとバイアスディファレンス
AA、AC、AG、AT、CA.....TTまで4x4で16通りの2塩基が考えられます。あるDNA配列についてその1番目から2塩基を調べます。続けて2塩基目から2塩基を調べます。これを配列の最後まで繰り返すことで、16通りの2塩基がそれぞれ何回登場したかを調べます。また相補鎖についても同様に調べ、表鎖と裏鎖の両方についての登場回数の総和を調べます。また、A、C、G、T、それぞれが何回登場したかを調べます(表鎖と裏鎖で)。このデータを基に計算を行います。例えばACの出現回数が18425回、Aの出現回数が64735回、Cの出現回数が102687回、調べた配列の総長が334844 base (つまりここで解析しているのは167422 baseの配列)であったとします。この時、2塩基「AC」のバイアス値は、
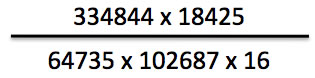
で計算されます。ACの登場回数を、A単独の登場回数とC単独の登場回数で除しているので、この値が大きいときは、AあるいはCの単独の登場回数の割に、ACの登場回数が多い(偏って多く出現している)と理解することができます。この計算式で、16通りの2塩基についてそれぞれバイアス値を計算します(裏鎖についても調べていますので、例えば「AC」と「GT」はそれぞれ同じ値を持つことになります)この16個よりなる数値のセットをバイアスデータと呼びます。ここで計算したのは、配列全体についてのバイアスデータです。
では次に、DNA配列中の一定の長さ(例えば1 kb)の部分について考えてみます。いま知りたいのはこの部分の2塩基組成が、配列全体の2塩基組成に比べてどれぐらい異なっているかということです。そこで、この1 kbの配列について上と同様にバイアスデータを算出します。これをローカルバイアスデータと呼びます。どれぐらい異なっているかを調べるには、16種類の値それぞれの間で引き算をして絶対値にした値の総和を計算します。この値をバイアスディファレンスと呼びます。この値が大きいほど、2塩基の組成が異なっていることを意味します。
DNA配列全体について、各部位のバイアスディファレンスを計算するには、1 kb程の範囲(これをwindow sizeと呼びます)についての計算を、500 baseぐらいづつずらして計算します(step sizeと呼びます)。
コアバイアスデータ
あるゲノムDNA配列についてバイアスデータを計算したとします。ゲノムDNA配列の中には外来領域も混ざっており、計算したバイアスデータはそれも含めた計算値です。GenoemMatcherのDinucleotideBiasSignatureでは、配列全体のバイアスデータに基づいて、配列の各部位のバイアスディファレンスを計算し、その値が低い方の半分について、バイアスデータを計算し直したモノをコアバイアスデータと呼んでいます。コアバイアスデータにどれぐらい意味があるかは未知数ですし、文献にもなっていません。
実行例
3,448,421 baseの環状のレプリコンの解析例を示します。
- 「解析するDNA配列の入力方法」として「ファイル」を選択し、DNA配列を一つ選択します。
- 「解析するDNAの形状」として「環状」を選択します。
- 「比較に用いるbiasデータ」として「ファイル中の配列のbiasデータ」を選択します。
- windowサイズを1000 bpに、step sizeを500 bpにセットします。
- 実行ボタンを押します。しばらく待ちます。
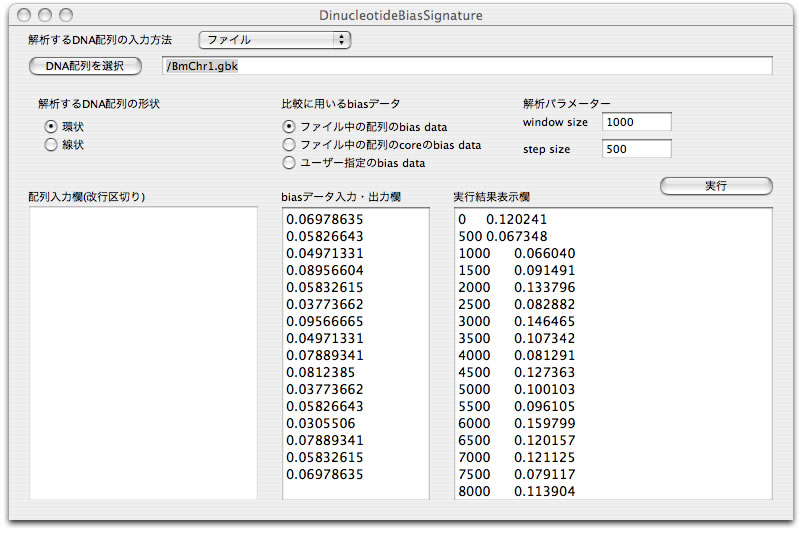
中央のテキストフィールドに示されているのは配列全体のバイアスデータです。右のテキストフィールドに示されているのが配列中の各部位のバイアスディファレンスです(表計算シートにペーストしてください)。
注意とヒント
「解析するDNA配列の入力方法」として「テキストフィールド」を選択した場合は、左側のテキストフィールドにDNA配列を改行区切りで入力して下さい。この場合、解析パラメーターに関係なく、入力配列一つ一つについて、バイアスディファレンスを計算します。また「比較に用いるbiasデータ」には「ユーザー指定のbias data」を指定し、中央のテキストフィールドにバイアスデータを指定形式に従って入力してください。
出力されるバイアスデータは、AA、AC、AG、AT、CA、CC、.....TTの順です。この形式はそのままユーザー指定のバイアスデータとして利用できる形式です。
「比較に用いるbiasデータ」として「ファイル中の配列のcoreのbiasデータ」を指定して実行したときに、中央のテキストフィールドに表示されるのは、コアバイアスデータです。
ArcWithColorにもバイアスディファレンスを計算させる機能がついています。
入力ファイルとしては、メイン機能と同様の形式が読み込めます。