このページは比較ゲノムソフト「GenomeMatcher」を有効にご利用いただくために、背景となる知識について解説しているページです。GenomeMatcherは比較ゲノムだけでなく、情報処理にも有用な機能を多く備えたソフトウエアです。
GenBank形式 / feature keyとqualifier/ FASTA形式 /multi FASTAファイル
ファイル名 / ファイルパスとフォルダパス / 改行コード / ファイル形式 / 拡張子 / コンテクストメニュー / スレッド / ベクターグラフィックス / ポイント / GUI / 強制終了 / スクリーンキャプチャ / テキストフィールド
ORFとCDS
MAFFT / MUMmer /CONSERV/ BLAST / bl2seq / blastallとformatdbとBLASTデータベース/ e-value/ HSP / クエリカバレージ / クエリ / サブジェクト / SNP /
しきい値
コンティグ / リファレンスゲノム
DNA配列とDNA配列に付された注釈(アノテーション)の記述形式の一つです。GenBank形式で記述されたテキストのファイルをGenBankファイルと呼びます。GenBankファイルはテキストエディットで開くことができます。もっとも汎用な形式である一方で、データの処理がしにくいという欠点があります。GenomeMatcherにはGenBank形式のファイルの中身を表計算シートで取り扱いやすい形式に変換する機能「ExtractFromGenBankFile」が付いています。またGenomeMatherの多くの機能ではGenBankファイルを入力ファイルとすることができます。

GenBankファイルの例。左側にある「source」「gene」「CDS」などをfeature Keyと呼ぶ。また右側にある「/」スラッシュから始まる文字列(locus_tag、product)などをqualifierと呼ぶ。
GenBank形式のデータでは、DNA配列中のどの部位に何があるかについて複数件が記述されています。その一件一件に1つのfeature keyが付けられています。例えば、CDS、tRNA、repeat_region、等であり、当該情報が何に関する情報かを示しています。1件のデータを見てみると、translation、locus_tag、note等と書かれています。これらはqualifierと呼びます。GenBank形式の項目も見てください。
ある配列の名前と、配列が以下のようになっている形式のこと。すなわち「>」記号に続いて「配列の名前」、「改行(\n)」、「配列」、となっている形式。配列はDNA配列あるいはアミノ酸配列。>から改行までの間(配列の名前の部分)をヘッダーと呼ぶこともあります。
 見えませんが、Bxe_B3028とMの間には改行キー(¥n)が入っています。
見えませんが、Bxe_B3028とMの間には改行キー(¥n)が入っています。
配列の名前と配列のセットを複数含むファイル。DNA配列とアミノ酸配列両方を同時に含むことはありません。
プログラムによっては日本語名のファイルを受け付けないことがあります。安全のため、ファイル名およびそれが格納されているフォルダ名についても半角英数字のみとすると良いでしょう。スペースも名前に含めない方が無難です。
パソコン中でファイルは階層的になったフォルダに格納されています。ファイルを指定するときには、そのファイルが一意に定まるように指定する必要があります。階層構造のてっぺん(ルートとも言いスラッシュ「/」で表す)から様々なフォルダを経由してファイルへといたる道筋を絶対ファイルパスと言います。任意のフォルダやファイルの絶対パスを調べるには、ターミナルを起動して開いたwindowにフォルダ/ファイルのアイコンをドラッグします。
よく使われる改行コードは3種類あります。3種類はそれぞれ、「\n」、「\r」、「\n\r」と表記されます。普段は意識しなくて良いのですが、意識しましょう。なぜならblast等が\nのみを改行キーとして認識するからです。作成している文書の改行キーを確認/変更するにはGenomeMatcherの付属機能「StringFormatter」を利用してください (ウェブページ上では円マークで表示されているかも知れませんが、実際にはスラッシュ「/」の傾きが反対になったバックスラッシュです)。
多くの場合、プログラムはRTF形式(リッチテキストフォーマット)を読み込めません。ファイルはtxt形式で保存するようにしてください。テキストエディットを利用する場合は、メニューから「標準テキストにする」を選ぶと、txt形式で保存できます。また多くの場合ファイル中の改行キーは\nである必要があります。逆に改行コードが¥rである必要があることはほとんどありません。詳しくはは改行コードの項目を見てください。
ファイルの名前中、最後にあるドット以降を拡張子と呼びます。「001.txt」がファイル名であれば拡張子は、txtです。Macでは拡張子を隠す設定があるのですが、隠してしまうと表示されているファイル名と実際のファイル名が異なることとなります。またターミナルなどでファイルを操作するときに、拡張子を省略することはありません。そのような訳で、拡張子を隠さない設定にすることを推奨します。
コントロールキーを押しながら左クリック、あるいは、右クリックすると、メニュー項目が表示されることがあります。このときこのメニューをコンテクストメニューといいます。
コンピューターが動作するときの処理進行の単位。4つのCPUを持つMacに1つの処理を任せると、4つのうち1つのCPUのみが利用されます。このとき他の3つは何もしておらず、パフォーマンスの向上には寄与していません。パフォーマンスを向上するには、4つのCPUに4つまたはそれ以上の処理単位を実行させると良い場合があります。
グラフィックスは2つに大別できます。1つは写真の様に絵のデータが各座標の画素データとして保持されているグラフィックス(ビットマップイメージとも言います)、もう一つは、「座標(0,0)から座標(10,10)に幅1の赤い線が引いてある」といったように描画に必要なデータが、情報として維持されているグラフィックスです。後者をベクターグラフィックスと呼びます。ベクターグラフィックスは、拡大してもぼやけない、画像を編集できる(線を消したり、線を太くしたりできる)といったメリットがあります。またデメリットとしては、あまりに描画情報が大きくなると描画に時間がかかるという点が挙げられます。GenomeMatcherでは基本的にベクターグラフィックスを出力できるようになっています。これは論文用の図画の作成にベクターグラフィックスの方が適しているからです。ベクターグラフィックスが重すぎて取り扱いづらい場合などは、スクリーンキャプチャすることでビットマップイメージに変換してください。
ベクターグラフィックスを取り扱える画像解析ソフトとして、筆者はCanvas Xを使っています (ファイルメニューの「開く」からPDFファイルを選択)。illustratorですと文字を文字として扱えなくなってしまうようですが、ファイルをAcrobatで開いてeps形式で保存し直すと、大丈夫なようです。
GenomeMatcherでグラフィックスを描画する際に使用する単位はポイントです。1インチ = 72ポイントです。フォント12ポイント、線幅0.5ポイントといったように普段から使っている単位です。なおA4の紙は横幅595ポイント、縦幅842ポイントです。
グラフィカルユーザーインターフェースを略してGUIといいます。インターフェースとは2つのモノの接点、ここではプログラムと人間の接点のことです。この接点がグラフィカルな場合、それをGUIと言います。画面上のボタンやテキストフィールドやプルダウンメニューなどのグラフィックスで表される画面要素を操作することで動かすソフトウエアは「GUIを持つ」とか「GUIツール」などと言います。対義語はキャラクターユーザーインターフェース(CUI)で、こちらはコマンドラインにコマンドをキーボードから入力して操作するようなインターフェースのことを言います。
Macのソフトを強制終了するには、「コマンド」+「オプション」+「escキー」を同時に押し、強制終了したいソフトを選択します。強制終了しても、内部的に実行されていたプログラム(blastall等)は強制終了されません。必要ならばアクティビティモニタを起動して、終了したいプロセスを選択して「プロセスを終了」ボタンを押します。
Macではプログラムの実行状況を確認するのに、「アクティビティモニタ」が使えます(Finderの「移動」→「ユーティリティ」メニューから「アクティビティモニタ」を選択します)。CPUの稼働率や、今何が動いているのかをある程度確認できます。またアプリケーションがどれぐらいメモリを消費しているのかを確認数することができます。
Macでは、「コマンド」+「シフト」+「コントロール」+「4」を押すとパソコンの画面をキャプチャするモードになります。カーソルの形が変わったところでキャプチャしたい範囲をドラッグすると、ドラッグした範囲の画像がペーストボードに記録されます。キャプチャした画像は、しかるべきところにペーストできます。画面のキャプチャをすると時に解像度が十分でなくなるときがあります。その場合、当該範囲をソフトウエア的に拡大してからキャプチャすると改善することがあります。
Macのソフトウエアでよく見られるテキストの入力欄の1つです。最初から数行の高さを持っているテキストフィールドがある一方で、1行しかないように見えて何行にも渡ってデータが入力されていることもあります。また横幅が十分でなく、文字列の右側が隠れてしまっている場合もあります。
テキストフィールドへの入力の終了は、リターンキーを押すか、別のテキストフィールドを選択することでプログラムに伝えられます。「あれ?設定がすぐに反映されない??」と思ったら、入力後リターンキーを押してみてください。
テキストフィールドの内容を間違いなく全部消去するには、テキストフィールドを選択後、セレクトオール(コマンド+A)したのちにデリートキーを押します。テキストフィールド内の内容全部を得るには、セレクトオール後、コピーして、どこかにペーストします。テキストフィールド中に、タブを入力するには「オプション」+「タブ」、改行を入れるには、「オプション」+「リターン」とします。この場合の改行コードは¥nです。
CDSとはcoding sequenceの略で、DNA配列のうち1つのタンパク質をコードする部分をCDSと呼びます。GenBank形式のfeature keyとしてもCDSが使われています。
ORFはopen reading frameの略です。「open」には妨げるモノがない、との意味があり、ORFは「妨げるモノがない読み枠」との意味です。「妨げるモノ」とはこの場合終止コドンとなりうるトリプレット(TAA、TGA、TAG; ここでは実際にタンパク質コード部分の終止コドンとなるかどうかに関係なく、これらトリプレットを終止コドンと呼びます)であると考え、ORFとは各読み枠における終止コドンから終止コドンの間の部分を指すと考えるのが良いようです。
すなわちそこにタンパク質がコードされていようがいまいが、コードされる読み枠であろうがなかろうが、開始コドンがどこにあろうが関係なく、終止コドンから終止コドンまでをORFと考えるべきです。
ドライ系の人がORFをこのように捉えている一方で、ウエット系の人は、沢山あるORFの中から実際にタンパク質をコードしていると考えられるORFの部分、すなわち実際にタンパク質をコードしていると考えられる開始コドンから終止コドンの部分(つまりCDS)をORFと呼ぶことがあり、時にタンパク質をコードする遺伝子にorf1、orf2などと名前を付けたりすることもあります。
GenomeMatcherのアノテーション支援機能にORFを抽出する機能がありますが、CDSとして同定されるには開始コドンの存在が必須であることをふまえ、ORFの中で最も上流にある開始コドンとなりうるトリプレット(ATG、GTG、CTG)から終止コドンまでを「ORF」と呼んで抽出しています。
配列のアラインメントツール。clustalWと比べて桁違いに速い。
DNAレベルであるいはアミノ酸配列レベルでアラインメントをつくるnucmerとpromerを主とするゲノムアラインメントツール。
配列間で100%保存されている塩基配列を同定するツール。ユーザー自身がソースコードをダウンロードし、コンパイルする必要がある。コンパイルしたプログラムを置いた場所を、GenomeMatcherの環境設定window中で指定することで利用可能になる。入手先: http://www.gen-info.osaka-u.ac.jp/~ngoto/CONSERV/
最もよく使われている相同性検索プログラムです。GenomeMatcherには、一般に良く使われているBLASTプログラム「blastall」と、BLASTプログラムの亜種であり2つの配列の比較に特化した「bl2seq」が入っており、状況に応じて2つのプログラムを使い分けています。どちらが動いているのかは必ずしもわかりやすくはなっていません(改善予定です)。どちらのプログラムについても環境設定等でパラメーターの詳細を設定することができます。ただしプログラムが必要とする設定を変更しようとしても、反映されないようになっていますので注意してください。
よく使うblastallのパラメータ
-m 8 テーブル形式で結果を出力します。
-v 10 トップ10件の1行descriptionを出力します。
-b 5 トップ5件についてアラインメントを出力します。
-e 0.01 e-valueが0.01以下のヒットについて出力します。
-W 21 ワードサイズに21を指定します(このとき検出されるHSPには、21bp以上の連続した100%一致する部分が少なくとも一カ所あります)。数字が大きいほど、解析が早く終わる一方で、検出されなくなるHSPが増えます。primerの配列や新型シーケンサーのような短い配列をblastにかけるときには留意する必要があります。
BLASTプログラムの亜種であり2つの配列の比較に特化したプログラムです。GenomeMatcherのメイン画面のcompareボタンを押したときに使われるのはbl2seqです。2つのDNA配列ファイルを入力として受け付けるこためにデータベースを作らなくて良いこと、また配列中の比較する位置を指定できることから、コマンドラインからプログラムを実行するのが楽です。
bl2seqが2つのDNA配列を入力として受け付けるのに対して、blastallの実行時にはあらかじめ作成したBLASTデータベースが必要となります。blastallは指定したBLASTデータベースからクエリ(問い合わせ配列)と相同性を示す配列を検索します。blastallはNCBIのウエブページなどからダウンロードして自分のコンピューター上で動作させることもできます。この際、BLASTデータベースを自分のパソコン中に用意する必要があります。BLASTデータベースはNCBIのページからダウンロードすることもできますが、BLASTプログラムと一緒に配布されているプログラム「formatdb」を利用することで、任意の配列を含むmulti FASTAファイルを基にBLASTデータベースを作成することができます。GenomeMatcherには、BLASTプログラムであるblastallとformatdbプログラムが埋め込んであり、ユーザーの目に見えないところでformatdbやblastallが実行されています。付属機能の「MassiveBLAST」の中にはformatdbのGUIがあり、それを利用するとBLASTデータベースの作成が容易です(MassiveBLASTマニュアル)。
BLASTで解析する時に見いだされたヒットがどれぐらい意味のあるヒットであるかに関する指標です。0に近いほどより意味のあるヒットであることを示します。同じクエリであれば、データベースから見いだされたsbjectについて、データベースが大きいほどe-valueは大きくなり、データベースが小さいほどe-valueは小さくなります。
BLASTの用語。ある配列の一部分が、別の配列のある部分に似ているとき、この2つの部分配列のアラインメントを作ることができます。アラインメントを作り得た部分配列2つのペアをHigh-scoring Segment Pair(HSP)と呼びます。進化的に少し離れたDNA配列を比較すると、もはやアラインメントがつくれないほど配列が分岐してしまった部分が出てきますので、BLASTを実行することで沢山のHSPが作られることになります(アミノ酸配列に付いても同様のことが起こりえます)。
アミノ酸配列をクエリにしてblastp検索を実行すると、いろいろな相同配列が見つかります。クエリの全長にわたって相同性が見られる場合がある一方で、クエリのほんの一部にしか相同性が見られない場合もあります。クエリの長さに対して相同性が見られた範囲のアミノ酸配列長をクエリカバレージと呼び100分率で表します。例えば100アミノ酸よりなる配列をクエリにして、そのうち60アミノ酸の部分に他の配列との相同性が見られた場合、クエリカバレージは60%です。
問い合わせ配列のこと。BLAST検索のときに、データーベースに対して検索される配列のことをクエリと言います。
クエリをデータベースに対してBLAST相同性検索したときに、データベース中から見いだされた相同配列のこと。BLASTの出力結果では(おそらくqueryと文字数をそろえるため)sbjectと記されているため、本マニュアル中ではsbjectと表記することもあります。
一塩基多型のこと。すなわち同種のあるいは近縁の生物よりなる集団ついて、ゲノム中の特定のDNA配列に関して、いろいろな型(A、C、G、またはT)が見られるときこれをsingle nucleotide polymorphismと呼びます。だたしGenomeMatcherでは2つの配列を比較したときに塩基配列に違いが見られた1塩基のことをSNPと呼んでおり、厳密にはSNPではありません。SNPはMUMmerにより検出していますが、MUMmerの方でこれをSNPと読んでいるのでそれに倣いました。
数値がある基準を満たしたときに意味があると考えるときに、基準となる値のことをしきい値(閾値)と呼びます。例えばアミノ酸配列の相同性が60%以上ある時に有意な相同性であると考えるとき、60%がしきい値です。
例えばあるゲノム配列について、1-1000 baseの範囲のGC含量を調べ、次いで500-1500 baseの範囲のGC含量を調べ、というように一定の長さに渡る解析を、解析範囲を少しずつずらしながら行って全体を調べる時、この一定の長さをwindow sizeと呼び、ずらす長さをstep sizeと呼びます。
ショットガンシーケンシングによって得られたたくさんのリードについて、配列の同一性を頼りに配列を重ね合わせ(アッセンブルした)ときにできる配列。
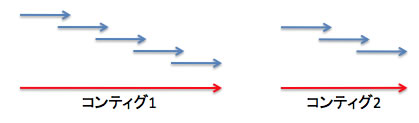 リード(青)とリードを重ね合わせてできたコンティグ(赤)
リード(青)とリードを重ね合わせてできたコンティグ(赤)
あるゲノムを決定しているときに、その類縁ゲノムの配列が既に決まっている場合、それを参照しながらコンティグ間の関連を予想することなどができる。この場合参照対リファレンスゲノムと呼ぶ。