BLASTinterface
ローカルな環境で(ユーザのパソコンの中で)BLASTを実行するための機能です。BLASTデータベースをご自分のパソコン中にお持ちであれば、それに対してBLAST検索を実行することができますが、配列データを含むファイルを指定して実行することもできます(BLASTデータベースが内部的に作成されます)。またBLASTの結果を表計算シートに適合する形に変換する機能がついています。
はじめに
自前のデータベースに対してblast検索を実行するとなると、ウエブ上の検索サービスに頼るわけにいきません。BLASTinterfaceは、お使いのMac上でBLAST検索を実行するためのGUIです。お使いのMac中に持っているBLASTデータベースを指定してBLAST検索を実行できるほか、DNAやアミノ酸配列を含むファイルを指定してこれに対してBLAST検索を実行できます。また表計算シートで1行1データでアミノ酸配列を管理している場合、そのアミノ酸配列をblast検索したヒット結果を、配列の隣に貼り付けられるように整形する機能もあります。
クエリの指定
プルダウンメニューから「ファイルを指定」あるいは「テキストフィールドに入力」のいずれかを選択してください。ファイルは1件のみ指定できます。ファイル形式はGenBank形式、multi FASTA形式、テキスト形式のいずれかです。クエリを「テキストフィールドに入力」するときは、multi FASTA形式で入力してください。クエリ1件のみの時は配列データだけでも大丈夫です。
データベースの指定
プルダウンメニューから「指定ファイルをデータベース化して利用」あるいは「既存のデータベースを利用」のいずれかを選択してください。
「指定ファイルをデータベース化して利用」する場合、ファイルは複数指定できます。GenBank形式のファイルの場合、使用するプログラムによってDNA配列あるいはアミノ酸配列が抽出されて(内部的に)データベース化されます。
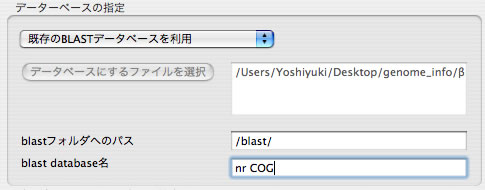
「既存のデータベースを利用」する場合は、データベースの置き方に2つのルールがあります。第1に、お使いのMacのどこかに「blast」という名前のfolderがあり、その中の「db」フォルダの中に利用したいデータベースファイルが入っていなくてはなりません。例えばMachintosh HDの直下にblastフォルダを置く場合は、/blast/db/<database files>となっている必要があります。第2に、このblastフォルダへのパスを、設定画面上で指定しなくてはなりません。この2つのルールを守っていただけると、スペース区切りで複数のdatabaseを指定することができます。下図は2つのデータベース、nrとCOGを指定したところです。blastフォルダへのパスのところで、「blast」のあとにスラッシュを入れるのを忘れないでください。
プログラムとパラメーターの設定
プログラムはプルダウンメニューより選んで下さい。パラメーターはスペース区切りで入力して下さい。
結果の処理方法
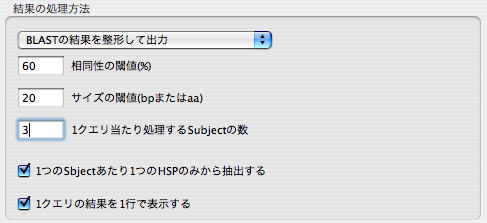
BLAST検索の結果に、何も手を加えないで表示するにはプルダウンメニューより「BLASTの結果をそのまま出力」を、整形して表示するには「BLASTの結果を整形して出力」を選んで下さい。後者は表計算シートに出力結果をコピーペーストするためにある機能です。
「BLASTの結果を整形して出力」する際に、出力する情報についていくつか制限を設けることができます。
- 相同性のしきい値。相同性の低いヒットを無視します。
- サイズのしきい値。短いヒットを無視します。
また1つのクエリ当たり何件までのヒットを処理するかを指定できます(0を指定すると全件処理します)。top 3までなら3を指定します。
「1つのSbjectあたり1つのHSPのみから抽出する」にチェックを入れると、あるクエリがあるsbjectの2箇所以上にヒットする場合、1件目のみが取り扱われます。
「1クエリの結果を1行で表示する」にチェックを入れると、1つのクエリの結果は、1行に収まるように表示されます。
「BLASTの結果をそのまま出力」した場合、「BLASTの出力を解析」ボタンを押すことで、解析結果を整形することができます。
使い方の例
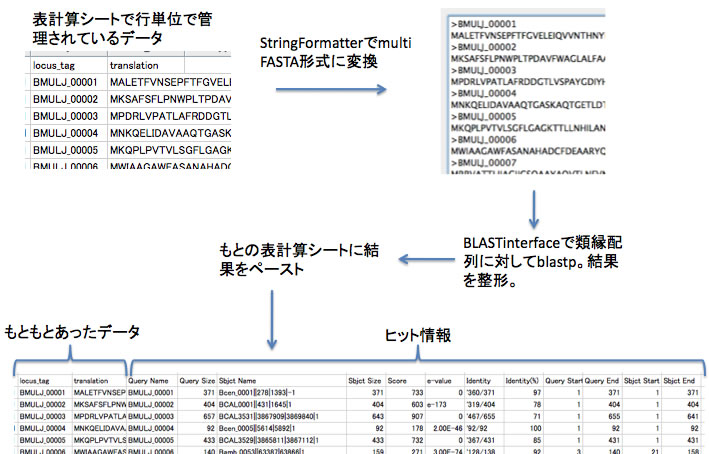
あるゲノムにある全CDSのアミノ酸配列を、表計算シートに持っているものとします(GenBank形式のファイルからアミノ酸配列などを取り出すにはExtractFromGenBankFileをどうぞ)。これを類縁菌のゲノムにコードされるアミノ酸配列と相同性検索をかけた結果を、アミノ酸配列の右隣に貼り付けた例を示します。ここではトップヒットのみ表示しています。
注意とヒント
GenBankファイルから読み込まれたアミノ酸配列は、データベース化に先立って内部的にmulti FASTA形式に変換されます。このとき各配列のヘッダーはローカスタグ、遺伝子名、開始位置、終了位置、向き、をパイプ記号(|)でつないだものとなります。GenBank形式ファイルの中のDNA配列が使われるときは、配列のヘッダーはファイル名となります。
入力した配列や指定したデータベースが、DNA配列なのか、アミノ酸配列なのかを識別する機能はついていません。使用するプログラムを間違えないように気を付けて下さい。
例えば表計算シートに、1000行に渡って1行当たり1つのアミノ酸配列とその配列名が記入されているとします。これをStringFormatterを利用してmulti FASTA形式に変換します。これをクエリとして、何らかのデータベースに対してblastpを実行した結果を整形することを考えます。このとき、上の図のように設定して得られた結果は、この表計算シートの配列の横に貼り付けることができます。このときデータは行方向にずれずに、トップ3件目までの情報が表示されることになります。データがずれないのは、ヒットがない場合でも1行空行を挟む仕様になっていること、2件目以降のヒットを改行せずに表示する仕様になっていること、によります。
別の手段でblastを実行したテキストデータをお持ちの場合、右側のテキストフィールドに貼り付けて「BLASTの出力を解析」ボタンを押すと解析できるかも知れません。
「指定ファイルをデータベース化して利用」する場合、実行ボタンを押す度にファイルの中身を読んでデータベースを作成します。指定ファイルが多い場合などは時間がかかりますので、同じデータベースに対して反復して解析を実行するときなどは、先にデータベースを作成し「既存のBLASTデータベースを利用」した方が早いかも知れません。MassiveBLASTにデータベースの作成を支援する機能がありますのでご利用下さい。
version1.350で仕様が変更になっています。更新をお願いします。