ContigAligner
複数のコンティグを、リファレンスゲノムに対して貼り付ける機能です。隣接するコンティグを予想することができます。またコンティグを2つ(あるいはもっとたくさん)選択すると、そのコンティグ間のギャップを埋めるのに必要なprimer配列の候補を出力します。
はじめに
第2世代シーケンサーでゲノムを読んでアセンブルすると、多くの場合、1本の配列にすることはできず多くのコンティグが得られるものと思われます。近縁種のゲノム配列が既知であれば、得られたコンティグをそれぞれ貼り付けることでコンティグ間の位置関係を推測することができます。位置関係が推測できれば、コンティグ間のギャップを埋めるためのPCR primerセットをデザインできます。また、「コンティグ全体に対して3'末端がユニークである」primerがあれば出力します。このような3'端がユニークであるprimerを用いることで、ゲノムを鋳型としてシーケンシングを行い、リードデータを得ることができるかも知れません。
解析手順
最初は以下の手順に従って操作してください。<>内は、6000本のcontigs、8.5 Mbのリファレンスゲノム、各8Mbの類縁ゲノム3つをテストデータとして実行したときの、筆者のパソコンを用いて実行したときの待ち時間です(version 1.363での時間です)。
- コンティグ配列を含むmulti FASTAファイルを選びます<1.9 sec>。
- リファレンス配列を含むGenBankファイルを選びます(ただのDNA配列を含むファイルでも可)<7.2 sec>。
- 「contigs VS reference 」box中の「blastn」ボタンを押します (全contigを、リファレンスゲノムDNAに対してblastallのblastnで解析します)<1 min 12 sec> 。blastn終了後、blastnの結果を保存できます。保存しておけば次回以降「open blastn result」からこれを読み込むことができます。
- 「contigs VS contigs」box中の「blastn」ボタンを押します (全contigを、全contigDNAに対してblastallのblastnで解析します)<1 min 30 sec>。blastn終了後、blastnの結果を保存できます。保存しておけば次回以降「open blastn result」からこれを読み込むことができます。
- search primerボタンを押します。新しく開いたwindow中でパラメーターを設定して、「search」ボタンを押します<16 min 45 sec>。
- 「add other genomes」ボタンを押します。新しく開いたwindow中の「selsect genome」ボタンで、他の類縁ゲノム配列があれば選択します。必要なだけ類縁ゲノムを追加したら、「add genomes」ボタンを押します(bl2seqのblastn)<2つの類縁ゲノムを選択。56sec>。
ここまでで貼り付いたコンティグが図示されますので、表示位置や描画レートを変更するなどしてご覧ください。
表示例
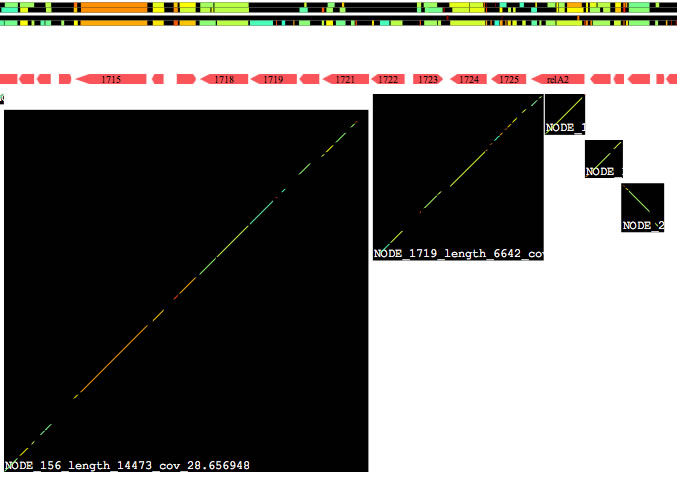
黒いパネル1つ1つがコンティグを表します(上図には5つのパネルがある)。
図の上部の4本の線(黒い線の上にいろいろな色の線が描かれている)の1番下は、各コンティグをリファレンスゲノムに対してblastnにかけたときに、ヒットが見られた部分を重ね合わせて表示したものです(「ヒットの総和」)。下から2番目は、リファレンスゲノムをそれ自身と比較したときの結果(ゲノム内の重複部位を示す)。上の2本の線は、類縁のゲノム配列をリファレンスゲノムに対してblastnをかけたときのヒットの様子(リファレンスゲノムのどの部位が近縁種で保存された部位であるか)を示します。リファレンスゲノムの注釈も表示されています。
パネルをコマンド+クリックすると、下図に示すグラフィックスが追加されます(version 1.365より)。これはcontigを灰色の矢印の場所にこの向きで置いたときに、検出される相同性を示しています。
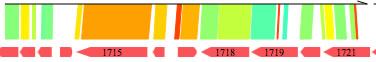
パネルの見方
各パネルの左下を原点として垂直方向にコンティグを配置したときに、検出される相同性が斜めの線で表示されています。パネルの縦の辺の長さはコンティグの長さを表します(比率を変えることができます)。パネルの中に見られる赤い水平線は、そのコンティグをコンティグ全部に対してblastnをかけた時にヒットが見られた部分を示します(ミスアセンブルしている可能性のある場所を示します)。
パネル横の辺はリファレンスゲノム中の比較対象になっている部分です。どこからどこまでが比較対象になっているかは、下の図の様になっています。2つのaは、そこが同じ長さであることを示します(bについても同様)。
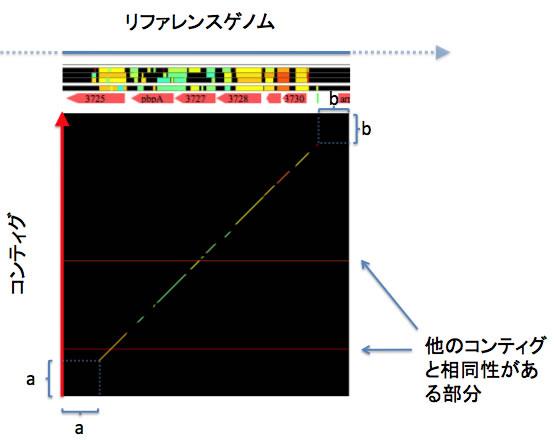
1つのコンティグ配列が、リファレンスゲノムの複数の部分と相同性を示した場合、複数のパネルが作られます。このような兄弟関係にあたるパネル間は、コンテクストメニューから行き来することができます。
リファレンスゲノムとゲノムを決めようとしているゲノム(ターゲット)のどちらか、あるいは両方に、特異的領域が存在するときについて、観察されるパターンをマニュアルの最後に示します。
primer配列
コンティグ間のギャップを埋めるには、隣り合っていると推定されるコンティグの隙間をPCR増幅し、PCR産物の塩基配列決定を行うことが考えられます。どのコンティグとどのコンティグが隣り合っているかを推定できない場合は、あらゆる組み合わせでPCRを行い、増幅したサンプルについて塩基配列を決定することができます。また別の方法としては、ゲノムを鋳型としてシーケンシングを行うことが考えられます。コンティグ間の関係を推定せずに実行できる反面、特異性の高いprimerと条件検討が必要です(我々が用いているプロトコールの詳細をマテメソに含む論文を近々投稿予定です)。
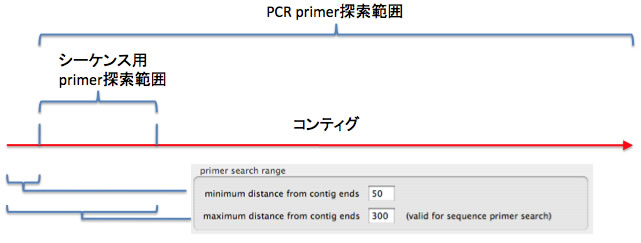
ContigAlignerはPCR用のprimerとsequence用のprimer(ゲノムを鋳型としてシーケンシングを行う用のprimer)をそれぞれ条件に従って探索します。primer配列は、全部のコンティグに対して一括して探すこともできますし、パネル1枚1枚について探すこともできます。新しく得られた塩基配列データを、既に得られている配列と重ね合わせるにはのりしろの部分が必要ですので、どちらのprimerもある程度コンティグの末端から離れた場所から探索する必要があるでしょう。
sequence用のprimerは3'端の特異性が高いこと、コンティグの末端からあまり離れすぎていないことが必要です。ContigAlignerは、primerの3'端の指定した長さが、全contigに対して特異的であるようなsequence primerを探索します。例えば11 merを指定した場合、出力されるsequence primerの3'末端の11 merは、他のcontig上のどこにも存在しません(すなわちユニークです)。
1つのコンティグに関して、5'方向に伸ばすprimerの探索範囲について下図に示します。sequence用のprimerは、限られた範囲が探索対象であること、3'端がユニークでなくてはならないこと、などから見つからない場合もあります。
primerの3'端の数塩基について、配列を指定することができます。以下に例を示します。
- S: 末端がGまたはCのいずれかである。
- SS: 末端の2塩基が、GG、GC、CG、CCのいずれかである。
- WS: 末端の2塩基が、AG、AC、TG、TCのいずれかである。
- WSS: 末端の3塩基が、AGG、AGC、ACG、ACC、TGG、TGC、TCG、TCCのいずれかである。
塩基コードとしては、W (A/T), S (G/C), R (A/G), Y (C/T), K (G/T), M (A/C), B (C/G/T),D (A/G/T), H (A/C/T), V (A/C/G), N (A/C/G/T)が利用できます。
その他の設定可能なパラメーターに付いては、実際の設定画面をご覧下さい。
コンティグ間をブリッジするprimerの出力
各コンティグの位置関係を推測する機能はついていません。すなわちコンティグ間の並びはユーザー自身で判断する必要があります。リファレンスゲノムに対する貼り付き方から、いくつかのコンティグの並びが推測できる場合に、その間をPCR増幅するためのprimerを出力するには、以下のようにします(すでにprimerをサーチ済みであるとします)。
- コマンドを押しながら、パネルをクリック。
- 続けて、隣り合うと考えられるパネルをコマンド+クリック。
- 隣り合うことが推測できるパネルが3つ以上ある場合は、順番に、コマンド+クリック。
- 「primers to bridge contigs」ボタンを押す。
- 1つのギャップについて2本のPCR primerが出力されます。
2つ以上のパネルを選択すると、選択されたパネルが薄いグレーの線で連結して表示されます。「delete」キーを押すと、選択が一つずつ解除されます(version 1.365以降では、各コンティグを、横向きに置いたときに検出される相同性が出力されます)。
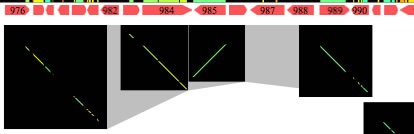 4つのパネルを順に選択した様子。
4つのパネルを順に選択した様子。
相同部分にジャンプ
任意のDNA配列あるいはアミノ酸配列を用いて、リファレンスゲノム中の相同配列が存在する部分へジャンプすることができます(「CompareSequences」の「search and focus」と同様の機能です)。
リファレンスゲノム中の相同配列の部分にジャンプする手順は以下の通りです。
- 「search and focus」ボタンを押します。
- 画面左側の設定をします。
- 検索に使うプログラムをプルダウンメニューから選択します。
- blastpで探す場合、見つかったCDSに付けるカラーコードを選択します(省略可)。
- クエリを入力します。
- 何番目のヒットに着目するかを入力します。1ならtop hitに、2なら2nd hitに、3なら3rd hitの部分にジャンプします。
- 「search」ボタンを押します。
同様に、コンティグに対して探索し、ヒットが見られたコンティグの部分へジャンプすることもできます。この場合、画面右側で設定をします。ほとんど上述の設定と同じですが、blastpで探索できない点、contigの名前で検索(完全一致のみ)ができる点、が異なります。見つかったコンティグには赤枠が付きます。
パネルのコンテクストメニュー
パネルにはコンテクストメニューが設定されています。
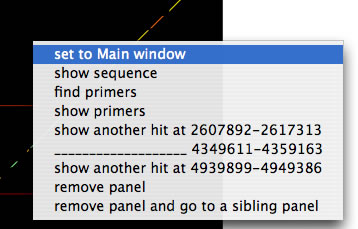
set to Main window <パネルのコンテクストメニュー>
メインウィンドウに、パネルの解析範囲をセットします。詳細な解析が可能です。
show sequence <パネルのコンテクストメニュー>
パネルのコンテクストメニューの「show sequence」を選ぶとコンティグの配列が表示されます。出力データは3つの部分からなっており、上から、1)このコンティグをリファレンスに対してblastnをかけたときの結果、2)1のうち、このパネルと関連のあるヒット、3)コンティグを全コンティグにたいしてblastnをかけたときの結果、です。いずれもヒットが見られた部分を色で示しています
find primers <パネルのコンテクストメニュー>
コンテクストメニューを呼び出したパネルについて、primerを検索します。既に設定されている条件での検索が開始しますので、あらかじめ設定window中で条件を決定しておいてください。
show another hit at ~<パネルのコンテクストメニュー>
1つのコンティグに関して、リファレンスゲノムの複数の場所にヒットが見られた場合、複数のパネルが作られています。このメニューから「兄弟」パネルに移動できます。ジャンプ先のパネルには赤い枠線が付き、表示範囲の中央に配置されます。
アンダーバーに続けて位置が書いてあるのは、呼び出し元のパネルです(選択すると自分自身にジャンプします)。
remove panel and go to a sibling panel ~<パネルのコンテクストメニュー>
コンテクストメニューを呼び出したパネルを取り除いて、「兄弟パネル」にジャンプします。
注意とヒント
10 Mb程度までの細菌ゲノムを解析するように作成しました。もっと大きな場合はうまく動作しない可能性があります。
ヒット長が短い場合は、リファレンスゲノムに貼り付かないようになっています。短いリードをそのまま貼り付けることはできません(「contigs VS reference」ボックス内の「ignore blocks shorter than ~」の後の数値が、どれぐらい短いヒットブロックを無視するかの設定です)。
パネルが作られないぐらいの短いヒットについても、「ヒットの総和」を表す線には示されています。
十分近縁な全ゲノム配列がない場合、本機能は効率良くご利用いただけません。
blastallやbl2seqのパラメータは、環境設定画面で変更できます。
本機能には、コンティグを分割したり、新たに得られた塩基配列データをもとにコンティグを連結する機能はついていません。ギャップを埋めるなり、コンティグの配列を伸ばしたりした場合は、あらためてコンティグ配列を読み込んで下さい。
出力されるprimerはあくまでもprimerの候補であり、PCR増幅やゲノムを鋳型としたシーケンシングが成功することを保証するものではありません。
ご要望をお寄せ下さい。
<パターン集>単純化した例を参考に示します。ターゲットは今からゲノムを決めようとしている生物のゲノムです。現時点では明らかになっていません。実際に見えるのはリファレンスゲノムと、それに張り付いたパネルだけです。「ターゲットへの挿入」は、「リファレンスからの欠失」かもしれませんがここでは区別していません。遠縁の配列をリファレンスとしている場合は、もっと複雑なケースが生じることに留意して下さい。
パターン 1: 最も単純な例。
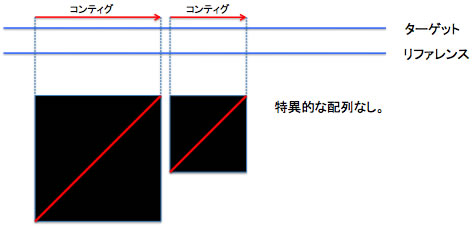
パターン 2: ターゲットに挿入がある場合1。
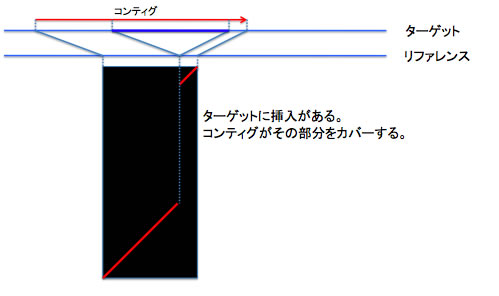
パターン 3: ターゲットに挿入がある場合2。パターン5と区別がつかないかもしれない。
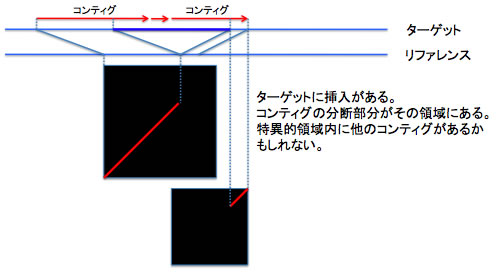
パターン 4: リファレンスに挿入がある場合。
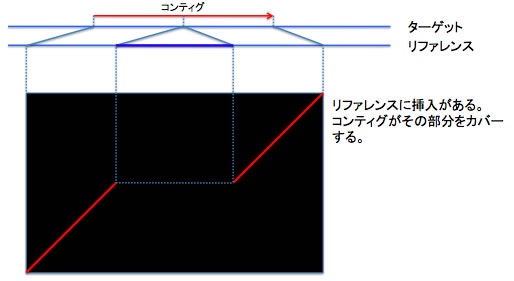
パターン 5: ターゲットにもリファレンスにも挿入がある場合1。パターン3と区別がつかないかもしれない。
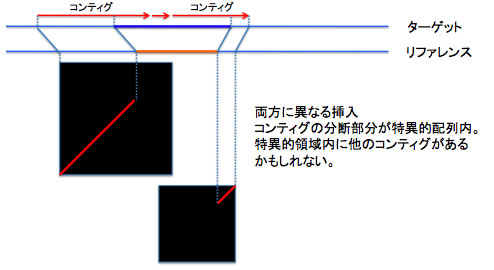
パターン 6 : ターゲットにもリファレンスにも挿入がある場合2。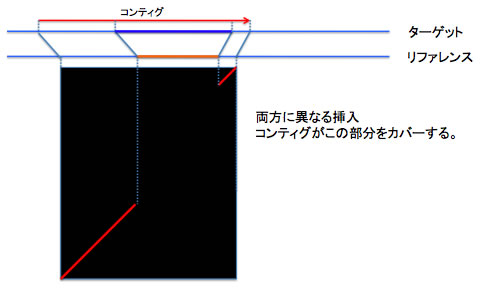
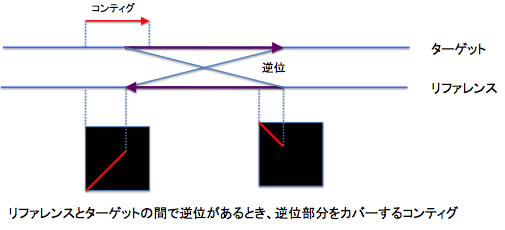
パターン7: ゲノムを決めようとしている配列と、リファレンスゲノムの間で逆位がある場合。