<補足 1>newblerの出力するコンティグと、コンティグをつなぐリードについて
ゲノム中のある部分の配列がゲノム中の他の場所にも存在する場合があります。このような領域をリピート領域と呼びます。newblerはリピート領域を1つのコンティグとして出力します。このとき、リピート領域とその隣の領域の両方にまたがるリードは、切り分けられて、リピートコンティグとその隣のコンティグへと帰属させられます。「コンティグをつなぐリード」とは、2つのコンティグの片方にリードの前半が、もう一方に後半が、属するリードのことを指します。「2つのコンティグがつながっている」ことは「2つのコンティグに帰属されたリードが何本かある」ことと同義です。GenoFinisherでは、リードによってつながった2つのコンティグの間には赤線がひかれます。またその線の太さは、そのリードの数に比例します。あるゲノムについて十分なカバレージでリードデータがある場合、基本的にはほとんどのコンティグの両方の末端それぞれが、すくなくとも一つのコンティグとつながっているはずです。このコンティグの序列を全て明らかにできれば、Finishingの重要な部分は終了すると言えるかと思います。
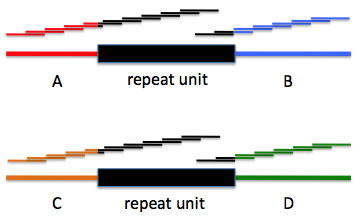
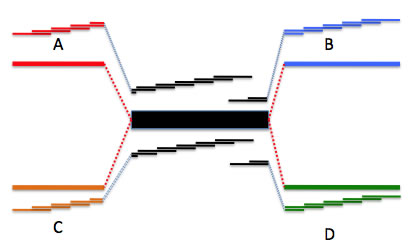
ゲノム中に2コピーのリピート領域がある場合。領域Aは領域Bと、領域Cは領域Dとリピート領域を介して繋がっている。この図のようにシーケンスリード長がリピート領域に比べて短い場合、AからBにつながっているのか、AからDにつながっているのかは、リード情報だけから知ることはできない。newblerはこのような場合、領域A、B、C、Dとリピート領域をそれぞれコンティグとして出力する。この際、例えばコンティグAとリピートコンティグ両方に含まれるリードは、切り分けられて、それぞれの領域に帰属させられる。ここでは切り分けられたリードを青い破線でつないで示した。コンティグ間をつなぐリードの数は454ContigGraph.txtファイルに記述されている。
<補足 2>イルミナのメイトペアペアエンドのリードの使用方法について
イルミナのメイトペアペアエンドデータを利用してスキャフォールド中のgapを埋めることができます。GenoFinisherのこの機能を利用するには、リードデータを適切に前処理する必要があります。ここでは、GenoFinisherで読み込むのに必要なデータ形式に付いて説明します。まず最初に必要な形式を示した上で、その形式を作成する方策について記述します。
- 1. イルミナのメイトペアペアエンドデータは、おおよそ最大20 MB程度のファイルサイズのファイルにmulti FASTA形式で格納されている必要があります。ファイルは(メモリ次第ですが)いくつあってもかまいません。これらファイル群は、「pairReads」フォルダに格納されている必要があります。またこのフォルダにはそれ以外のファイルが存在してはいけません。
- 2. 各ファイルには、ペアとなるリードが格納されている必要があります。ファイル1にリードの片方が、ファイル2にそのリードのペアが格納されているというのは不可です。
- 3. リードのIDは、IDの末尾が1で終わるものと2で終わるものがペアと見なされます。
- 4. 解析では、コンティグ末端に100%でマッチするリードが検索されます。つまりードは全長にわたってある程度のクオリティを有する必要があります。すなわち、低クオリティ部位を切り捨てる処置が必要です。
- 5. ペアとなるリードは、お互いに向かい合う向きである必要があります。イルミナのメイトペアペアエンドでは、得られる2つのリードはお互いに離れて行く向きですので、配列のリバースコンプリメントが必要です。
以下は、5の処理を終えたイルミナのリードデータが、2つのファイルに格納されている場合についてのデータ処理方法です。2つのファイルの同じ行番号に、ペアとなる配列が格納されているものとします。
- (i) ターミナルのpasteコマンドを使用して、2つのファイルを結合する。実行例: paste file1 file2 >pastedFile.txt
- (ii) perlのスクリプト i2.plを実行する。実行前に、テキストエディットでi2.plを開いて、必要に応じてパラメーターを設定する。このスクリプトの実行によって、ペアとなる2つのリード両方について5'末端から指定したスコア以上のクオリティが指定した長さ以上にわたって続くペアが抽出されます。例えば、ペアとなる2つのリード両方について、5'末端からスコア25以上のクオリティが連続して36塩基以上続くペアが抽出されます。実行例: perl i2.pl pastedFile.txt
- (iii) できたファイルを、ターミナルのsplitコマンドを使用して、20万行程度ごとに分割する。実行例: split -l 200000 targetFile.txt
- (iv)できたファイルを、「pairReads」フォルダに移します。このフォルダは、他のnewbler由来のファイルを格納したフォルダに置いてください。
- (v)newblerのアッセンブルに、メイトペアペアエンドの配列を、ペアの関係であることを認識させて混ぜ込むには、(iii)でできたファイルに対してパールのスクリプトi4.plを実行します。実行前に、テキストエディットでi4.plを開いて、必要に応じて出力ファイル名を設定します。実行例: perl i4.pl targetFile.txt
i2.plおよびi4.plについてはお問い合わせください。