必要なファイル
GenoFinisherは、newblerの出力データをもとに各種の処理を実行します。newblerの出力データは1つのフォルダに入れた上でそのフォルダを面画面で指定してください。このフォルダには以下の3つのファイルが格納されている必要があります。
454ContigGraph.txt
454AllContigs.fna (注)
454AllContigs.qual
注: 454AllContigs.fnaには、名前からすればすべてのContigの配列が含まれていそうですが、実際にはある程度大きいコンティグの配列のしか含まれていません。newblerでアッセンブルを実行するときに、1塩基以上であれば配列を出力する設定にすると良いでしょう(Parametersタブの中の、Outputタブの中にある、All contgs thresholdの値を0に設定)。
ペアデータがあり、スキャフォールドに基づいてFinishingする場合は、さらに以下の3つのファイルが必要になります。
454Scaffolds.txt
454Scaffolds.fna
454Scaffolds.qual
ペアデータがある場合に、以下の2つのファイルがあれば、スキャフォールド中のgapのいくつかを(追加の実験なしに)、埋める機能が使用可能になります。
454PairStatus.txt
454TrimmedReads.fna (注)
注: 454TrimmedReads.fnaを出力するには、GS de novo assembler version2.5.3以降が必要です。またデフォルトではこのファイルは出力されません。出力するにはassembleするときにParametersタブの中の、Outputタブの中にあるチェックボックスをONにする必要があります。
ContigDeepAnalysis機能を利用して、重複コンティグのなかにあるバリエーションについて明らかにするには、以下の2つのファイルが必要です。
454Contigs.ace
454ReadStatus.txt
注: 454Contigs.aceはデフォルトでは出力されない場合があります。
また、これらのファイルは同一のフォルダに格納されている必要がありますが、このフォルダの中に「pairReads」と名前をつけたフォルダを作成し、その中にmulti FASTA形式のシーケンスファイルを複数個入れておくことができます。各ファイルには、ペアとなるリードが格納されている必要があります。各ファイルのサイズはおおよそ20MB以内としてください。
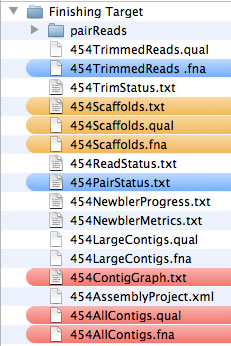
つまりこのようなフォルダ構成になります。d