GenoFinisherの機能
GenoFinisherの機能について概略を説明します。
- たくさんのコンティグをグラフィカルに表示する機能。繋がっているコンティグ間は赤い線で結ばれます。コンティグは動かして並べ直すことができます。
- 重複箇所を境にコンティグの繋がり方が不明になっている箇所について、自動で抽出し、コンティググラフを作成する機能。
- またこのとき、必要なPCRについての総合的な情報を出力する機能。
- コンティグの繋がりが明らかになった場合、その繋がりを指定することで、自動的にコンティグを連結する機能。
- コンティグの配列を編集する機能。クオリティスコアを維持しながら編集ができます。
- コンティグ内のクオリティスコアの低い箇所についてのレポート機能。
- コンティグをGUI上で連結する機能。
以下はスキャフォールドデータがある時に関係する機能です。
- スキャフォールド内の各ギャップ部分について、ギャップ前後のコンティグをつなぎうるルートの自動探索機能。
- また、発見したルートに関するコンティググラフを作成する機能。
- またルート確定作業に必要なPCRについての総合的な情報(後述)を出力する機能。
- 複数の可能なルートから1つを選択することで、ギャップにはまるDNA配列を出力する機能
- スキャフォールド内のギャップに、任意のDNA配列を挿入する機能。
- スキャフォールドの配列を、クオリティスコアを維持しながら編集する機能。
- コンティグ末端に含まれるリードと、そのペアとなるリードを抽出する機能。
- コンティグ末端の配列を抽出する機能。
- 上記2つの機能によって抽出された配列を、velvetまたはabyssでアッセンブルする機能(velvetおよびabyssを利用するにはユーザ自身でバイナリを入手していただく必要があります)。これによりGap配列が得られる場合があります。
またその他の機能として、以下のものがあります。
- multi FASTA形式のテキストデータ中でprimerを検索する機能。
- 任意のコンティグについて、ペアとなる2つのイルミナ配列の末端間の距離について、その分布を示す機能。
- 任意の配列について、multi FASTA形式のデータを指定することで、上と同様に末端間の距離の分布を示す機能。
- イルミナリードのフィルタリング機能
- 任意のコンティグを形成するリードを詳細に解析する機能(ContigDeepAnalysis)
コンティググラフ描画機能
コンティグ配列の編集機能
コンティグパネルのコンテクストメニューから「show sequnece」を選択すると、コンティグのDNA配列が、配列編集用のwindowへと送られ、表示されます。配列を編集後、その編集を呼び出し元のコンティグに反映させるには、「fix edited sequence」ボタンを押して下さい。このボタンを押すと、同名のコンティグパネル全てについて変更が反映されます。またprimerを新規に設定しようとすると、直ちに変更が反映されます。
この配列編集windowでできることは以下の通りです。
- 1. コンティグの基本情報を見ることができます。複数のコンティグの結合によって作られたコンティグについてはどのようなコンティグによって構成されているかをみることができます。
- 2. リーバスコンプリメントした配列を表示できます。
- 3. このコンティグを、コンティグ全体に対してblastnで相同性検索した時の結果を、各塩基の背景色として表示することができます。
- 4. クオリティが指定した値より低い塩基について、緑色の背景色で表示することができます。
- 5. 指定配列を検索して見つかった配列に指定した色を付けることができます。指定配列とそれの相補鎖の両方が、コンティグ配列全体に対して検索されます。
- 6. 指定位置にジャンプすることができます。
- 7. 指定範囲を選択することができます。
- 8. 指定した配列で、選択範囲を置き換えることができます。この際、置き換えた配列についてはその全ての塩基に付いてクオリティスコア99が付されます。選択範囲の配列を消去するには、空文字で置き換えます。
- 9. sequence用primerにはアンダーラインがひかれます。またPCR primerは青または赤で表示されます。青はF primer、赤は、R primerです。
- 10. 選択範囲について、その選択範囲の塩基番号が表示され、またこのときその配列のTm値が表示されます。
- 11. 選択範囲について、その部分をprimerとして設定することができます。primerの向きについて現在表示されている配列に対して右向きか左向きかで対応するボタンを押して下さい。この際、変更は全ての同名のコンティグに対して反映されます。
- 12. クオリティスコアについて、そのスコア分布を出力することができます。
- 13. クオリティスコアについて、指定した値以下のスコアの塩基の位置を出力することができます。
- 14. クオリティスコアについて、全部の塩基に付いてそのスコアを出力することができます。
スキャフォールド配列の編集機能
スキャフォールドパネルのコンテクストメニューから「load sequnece to viewer」を選択すると、スキャフォールドのDNA配列が、配列編集用のwindowへと送られ、表示されます。変更を反映するには「fix edited sequence」ボタンを押して下さい。コンティグの配列の編集windowとはできることに多少の違いがあります。
- blastnの結果は表示できません。
- リバースコンプリメントした配列は表示できません。
- primerを設定できません。
- プルダウンメニューから、ジャンプしたい場所にジャンプすることができます。
スキャフォールドの中のNNNNの部分にあてはまる配列を、このwindow中で手作業で置き換えることもできますが、ウィンドウ「Contig Graph(Task2)」中にある機能を使って挿入することができます。
primer総合情報出力機能
ペアデータがない場合は、ウィンドウ「Contig Graph (Task1)」を見てください。またペアデータがある場合はウィンドウ「Contig Graph (Task2)」を見てください。
ペアデータがない場合、重複コンティグを境にしたコンティグのつながり方をPCRで決めていくことになります。この場合、Contig Graph (Task1)ウィンドウに解決すべき課題が複数のシートに表示されます。
ここで、PCRにより解決すべきと判断される課題については、左上のチェックボックスをそのままにします(解決しなくても良いと思われるものはチェックを外します)。自動抽出されたすべての課題について、PCRによる解決を試みるかどうか、意志決定します。次いで、画面左下にある情報をエクセルシートに貼付けます。
このprimer総合情報は4つのパートから構成されています。
- パート1: primerの名前、配列、どのプレートのどの番地に分注するかについての推奨。
- パート2: プレートフォーマットで、1)各wellに入れる2種のprimer名、2)予想増幅産物サイズ、3)予想増幅サイズが見られたときのコンティグルート、4)課題シート番号。プレートフォーマットについては、PCRの予想増幅産物サイズに応じて3通りが準備される。
- パート3: 孤独末端間を総当たりでPCR増幅するためのPCR plate. 増幅サイズは不明なので「?」が表示されている。
- パート4: 各primerセットによる増幅配列、コンティグルートおよびルート中のコンティグの向きなど。
ペアデータがある場合、ウィンドウ「Contig Graph (Task2)」に必要な情報が出力されています。まずは「gapの配列を決定する機能」によりgapの配列決定を試みてください。これが不可能であったgapについてはgap前後にあるコンティグにアニーリングするprimerをデザインし、PCR産物を鋳型にシーケンス反応を行ってgapの配列決定を行う必要があります。上と同様に、PCRでの配列決定を行うgapについてのみチェックボックスをオンにし、画面左下にある情報をエクセルシートに貼付けます。
なお、primerの自動配列設定に失敗したコンティグについての情報が、「run」終了後に表示されます。必要に応じてマニュアルでprimerを設定してください。
日本遺伝子研究所(http://www.ngrl.co.jp/)は、合成DNAを指定した方法で混合して納品してくれました(混合しないものも)。primerを間違いなく2つずつ混ぜる操作はかなり大変ですが、おかげでかなり楽をすることができました。ただし多少の追加料金が必要でした。問い合わせて見てください。
アッセンブル機能
GenoFinisherはアッセンブルプログラムであるvelvetおよびabyssのGUIを提供しています。ただし、著作権の問題からこれらのプログラムはユーザー自身が入手する必要があります。ここではアッセンブル用のwindowについて説明します。windowの上部にあるテキストフィールド(1)は、比較的長い配列を入力するためのものです。またその左下にあるテキストフィールド(2)は短いリードを入力するためのものです。どちらもmulti FASTA形式で入力してください。ただし、前者については「色付きの」テキストを入力できます(ヘッダー">"の色が重要です。後述)。この状態で「run velvet」あるいは「run abyss」ボタンを押します。
velvetに関しては、"use reads as paired"チェックボックスをONにしておくと、(2)のテキストフィールドにペアと認識されるヘッダーを持つ配列がある場合、それらがペアであるとしてアッセンブルされます。またこの際、ペア間の距離を指定するようになっています。またvelvet、abyssともに、(1)のテキストフィールド中の配列を含めてアッセンブルするかどうかをチェックボックスで指定できます。アッセンブルされた結果は(2)の下にあるテキストフィールド(3)に出力されます。
「color contigs」ボタンを押すと、(1)と(3)のテキストフィールドにある配列について、conservが実行されます。conservは100%一致を検出するプログラムです。実行結果に基づいて、(1)のテキストフィールドにある配列の一部分と同じ部分が(3)のテキストフィールドの配列にあると、(1)のテキストフィールドの配列の"色"が(3)のテキストフィールドの配列に塗られます。また、(1)のテキストフィールドの配列のうち、ヒットしない部分の背景がシアン色に塗られます。また、(3)のテキストフィールドの配列のうち、相同部分が検出されなかった部分の背景色がシアン色になります。色の付き方を眺めることで、どの部分がどの部分に対応するのかを比較的楽に知ることができます。
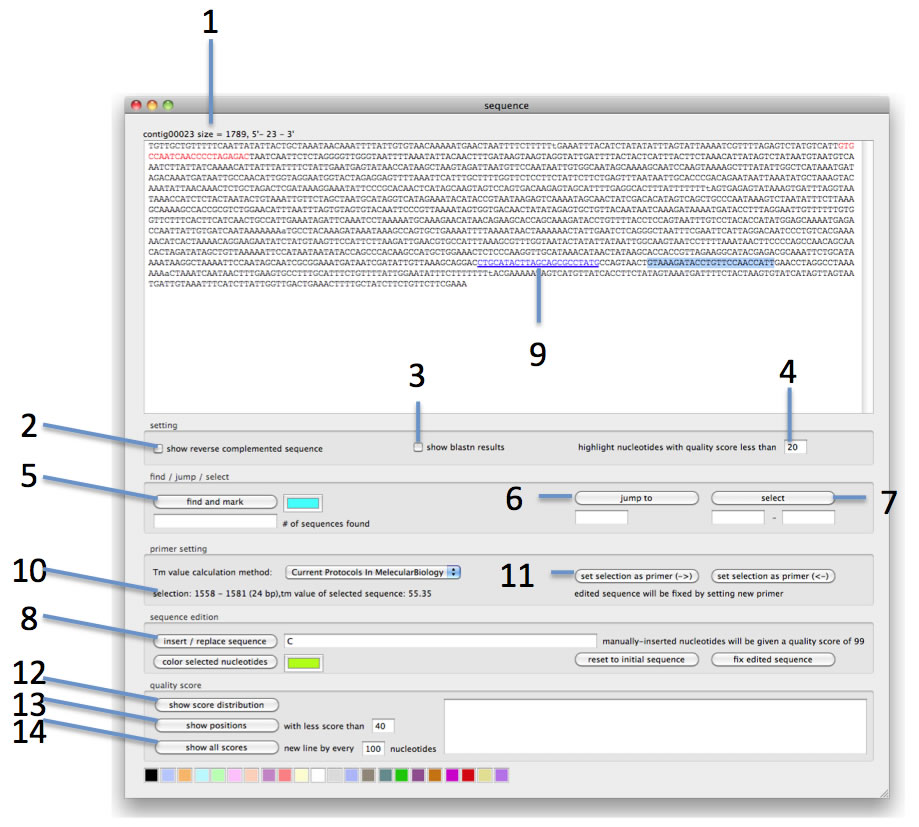
話は前後しますが、スキャフォールド内のコンティグ20の次が21であるとして、このgap相当のルートが一意に推定できている場合、コンティグ20のgapよりの配列、gapの配列、コンティグ21のギャップよりの配列が、それぞれ、赤色、緑色、青色で、テキストフィールド(1)に出力されるようになっています[gapの配列は必ずしも正しくないので(リピートコンティグであればバリエーションがある可能性がある)注意が必要です]。この3つの配列とあわせて関連するリードを混ぜ込んでアッセンブルしてできた配列について、配列のどの部分が元々コンティグ20で、どの部分がコンティグ21で、どの部分がgapであったのかを色によって知ることができます。

上の3つの配列と、イルミナのショートリードの配列(ここでは示していない)を混ぜ込んでアッセンブルしたのが下の配列。上の図の中の緑色の配列は配列が必ずしも正しくないと思われる配列(リピートの配列)。
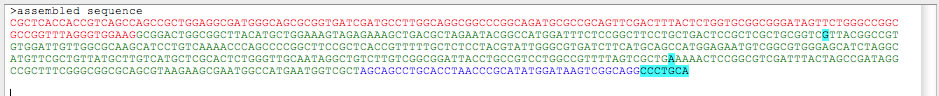
スキャフォールド中のコンティグ間のgap部分についてnewblerはNの連続として出力するようになっています。これはgap部分にリピートコンティグが存在していること、リピートコンティグにはバリエーション問題があること、によるものと推測されます。gapの配列が精度良く決まらない問題は、ペアデータがある場合は以下のように回避できる可能性があります。
- 1. gapの前のコンティグのgapよりの部分にヒットするリードを探す(図中赤矢印)。
- 2. 1で見つかったリードのペアを探す(図中緑矢印)。
- 3. 同様に、gapの後ろのコンティグのgapよりの部分にヒットするリードを探す。
- 4. 2で見つかったリードのペアを探す。
- 5. 必要に応じて、gapの前のコンティグのgapよりの部分の配列、gapの後のコンティグのgapよりの部分の配列を得る(図中青矢印)。
- 6. また必要に応じて、gap部分に相当するコンティグの配列を得る(ただしこれにはバリエーションに由来する配列上のミスがあるかもしれない)。
- 7. 以上の配列をあわせてアッセンブルする。
ペアとなるリードを利用してgap配列を決定する方法: イルミナリードの場合ユニークでないリード(黒矢印)そのものは出力されるが、そのペアとなるリード(紫矢印)は出力されないようになっている。
これにより、もしgapに相当する配列が得られたとすれば(特に5の配列を混ぜていなければ)、この配列にはバリエーションに由来する配列の間違いは含まれていないことになります。GenoFinisherは1から6の配列を簡単に収集できるようになっており、またアッセンブラー「velvet」と「abyss」のGUIとして機能します。なお、1および3についてはイルミナのメイトペアペアエンドデータに関しては、100%一致するリードについてのみ収集するようになっています。またこの際、100%一致したリードが他のコンティグの末端部分に100%でヒットするような場合には、対応するペアのリードを出力しないようになっています。また454のペアデータがある場合は、コンティグの末端部分を構成するリードIDを、ファイル「454PairStatus.txt」より取得し、その配列を、ファイル「454TrimmedReads.fna」より得ています。またこのペアとなるリードについても同様に配列をこのファイルから得ています。ペアリードの厚みが十分にあれば、追加の実験なしで、gapの配列を決定することができます。
multi FASTA形式のテキストデータの取り扱い機能
アッセンブル用のwindowでは、multi FASTA形式のテキストデータを取り扱う上で便利な機能が2つ付いています。一つは、(1)multi FASTA形式のデータの各配列を、全部同時にリバースコンプリメントする機能です。このとき、headderは全くそのままで配列のみがリバースコンプリメントされるので気をつけてください。もう一つの機能は、(2)各配列について5'方向および3'方向に伸長するprimerをデザインする機能です。このときのprimer検索条件は、「primerサーチとそのパラメーター」の節に記述したのと同等です。検索の前段階で、各配列はお互いにblastnで比較され、ヒットが見られた部分にはprimerがデザインされないようになっています(ヒットが見られた部分は灰色のバックグラウンドで表示されます)。また「primerサーチとそのパラメーター」の節に記述したのと同様に、2種類のprimerが検索されます。またデザインされたprimerの情報がwindow下部のテキストフィールドに出力されます。
(2)について例を示します。各配列について、2種類のprimerが検索されます(詳細は別に示します)。灰色の部分はblastnでヒットが見られた部分です。
リファレンスゲノムを使って各リードを分類します。GenoFinisherでは2通りの分類機能が実行可能で、1つは、100%一致するリードとしないリードに分類する機能、もう1つはblastnの結果に基づいてリードを振り分ける機能です(前者の方がずっと高速です)。
数GBのイルミナデータも、別のセクションで述べるフィルタリングをした後に、リファレンスに100%一致するかしないかで分類することでデータ量は数MB程度といった取り扱いやすいサイズになります。どのような解析をするかによって異なるかと思いますが、リファレンスに対して100%一致するリードがあまり重要ではないケースでは、100%一致するリードとそうでないリードに分類すると良いのではないかと思います。blastnによる分類では、各リードはblastnの結果に基づいて7区分に分類されます。
- 全長が100%一致するリードは[指定prefix]_perfectMatch.fnaに格納されます(区分1)。
- 全長がヒットし、またミスマッチの数が3塩基以内のリードは、[指定prefix]_lessThan4mismatchesに格納されます(区分2)。
- 全長がヒットし、ミスマッチの数が上記より多い場合は、[指定prefix]_queryCoverage100.fnaに格納されます(区分3)。
- リードの3'端にミスマッチが認められるが、それ以外の部分が100%一致である場合で、リード3'のミスマッチの長さが13塩基以下であるか、ミスマッチ部分に指定したアダプター配列がある場合は*1、5'の100%一致部分を、区分1に。またリード全長を、[指定prefix]_adaptorFoundAt3primeEnd.fnaに格納します(区分4)。
- リードの3'端にミスマッチが認められる場合で、5'のマッチ部分のミスマッチが3塩基以内であり、リード3'のミスマッチの長さが13塩基以下であるか、ミスマッチ部分に指定したアダプター配列がある場合は、5'の一致する部分を、区分2に。またリード全長を、[指定prefix]_adaptorFoundAt3primeEnd.fnaに格納します(区分4)。
- 以上のどれにも当てはまらないが、41塩基以上のヒットが見られるリードは、[指定prefix]_someSimilarityFound.fna(区分5)に格納されます。
- 以上のどれにも当てはまらない場合で何らかのblastヒットがある場合、[指定prefix]_no_significant_blastHit.fnaに格納されます(区分6)。
- 全くヒットがない場合、[指定prefix]_no_blastHit.fnaに格納されます(区分7)。
<その他のヒントと注意>
- multi FASTAファイルを処理することもできます。ただし、以下の2つの制約があります。(1)1リードが1行に納まっているmulti FASTA形式であること, (2)1リードの長さが255塩基以内
- 「k-merの出現頻度に基づいたイルミナデータのフィルタリング機能」を利用した後に、出力されたmultiFASTAファイル読み込ませることを推奨します。
- 各区分のリードを、なんらかのアッセンブラーでアッセンブルしたときに、複数本のリードが帰属するようなコンティグが作られる場合、帰属するリードには修正すべき箇所の情報が含まれていると考えられます。
- 既に指定ファイルがある場合は、「追加書き出し」されるようになっています。
- max threadおよびbatch sizeを変更することで、パフォーマンスが向上することがあります。
- まれに、振り分けが終了しているのに、プログラムがイベントループから出られなくなる不具合を発見しています。ご注意ください(修正中)。
k-merの出現頻度に基づいたイルミナデータのフィルタリング機能
イルミナリードのk-merの出現頻度に基づいて、リードをフィルタリングする機能です。この処理を行うことで、極端に出現頻度が低いk-merの部分と、極端に出現頻度が高い部分とを、取り除くことができます。つまり偶発的に読み間違えたと考えられる塩基や、アダプターの混入した部分などを取り除くことができます。ここではこの方法の原理についてk-mer=17 merとして説明します。
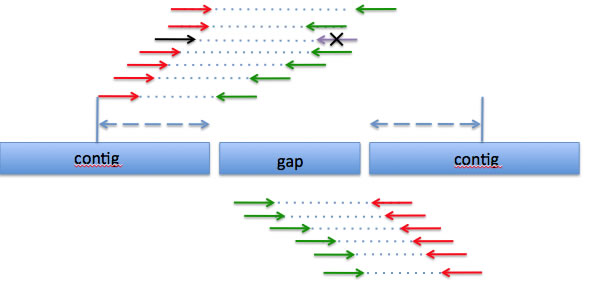
このフィルタリング処理ではまず最初に、イルミナリード全部のそれぞれについて、頭から17塩基ずつ取り出して、出現頻度を調べます。例えば1本の塩基長35のリードについては19通りの17 merが得られることになります。仮に5 Mbのゲノムがあり、シーケンスエラーなどが全くないとすると裏表も含めておおよそ1000万通りの17 merが得られます。ただし実際にはシーケンスエラーなどに由来するレアな17 merも相当ありますので、これよりはかなり多い種類の17 merが観察されることになります。
調べた出現頻度を、プロットすると下図のようになります。横軸は17 merが登場した回数、縦軸にはその回数登場したN-merの個数をプロットしています。例えばこの図では50回登場した17-merは44,136個あります。図に見るように、出現回数の低い(1回から数回)、17-merがかなりたくさんあることがわかります(この例では登場回数1回: 2,412,589、2回: 121.701、3回: 20484)。一度しか見られないような17-merがいったい何に由来するかについてですが、(1)リードの中にシーケンスエラーがある、(2)リードの末端がアダプター配列である、といったことが考えられます。また逆に、過剰に、例えば数万回も観察されるような配列は、実験操作で使用した配列、例えばアダプター配列などが読めているものと推測できます。
次いで、この出現頻度に基づいて、リードそれぞれについて、頭から17塩基ずつ取り出して、何回でてきた17-merであるかについて調べます。そうすると塩基長35のリードであれば、19個のスコア(数値データ)が得られることになります。このとき、エラーを含まないリードであれば、おおよそカバレージに近い値のスコアが19個並ぶことになります。エラーを含む場合は、そのエラー部位を中心として、小さい数値が見られることになります。例えば、5'端の1塩基が読み間違えている場合、19個の数値の1番目が低いスコアになります。また、スコアが極端に高い場所はアダプターなどシーケンスに利用したDNA配列であると考えることができます。
末端12塩基の出現頻度が低い例 (48塩基:32頻度データ): 0 0 0 0 0 0 0 0 0 0 0 0 80 76 77 76 76 77 75 75 75 75 75 79 77 78 78 81 77 77 80 80
配列の中央部分の出現頻度が低い例(50塩基:34頻度データ):90 90 90 88 88 90 89 93 90 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 139 138 138 139 139 136 134 135
GenoFinisherでこの機能を利用するには、「illumina read management」windowを開きます。このwindow中でイルミナファイルを選択後、「calcurate frequency」ボタンを押して、頻度解析を実行します。このとき、各リードについて、一定以上のクオリティスコアがある程度の長さにわたって連続する部分が頻度解析の対象となります。頻度解析が終了すると頻度分布データがファイルに書き出されていますのでテキストエディットなどでご覧ください(出力ファイルは、画面上部で指定したフォルダの中に、「指定prefix」_distribution_17merとのファイル名で出力されます)。なお「export frequency」ボタンを押すと、全N-merとその出現回数をファイルに保存してテキストエディットなどで開いて見ることができます。
頻度データが準備できたあとで、「export curated reads」ボタンを押すと、頻度データに基づいてすべてのリードについて(先頭から17塩基ずつ取り出しながら)スコア付けを行い、出現頻度が一定の範囲内(高すぎず低すぎず)に納まる部分の配列データが書き出されます(ファイル名は、「指定prefix」_curated_17mer、です)。なおこの際、リードのクオリティスコアは考慮されません。各リードについてどのようなスコアがついたかは、ファイルに出力されています(ファイル名は「指定prefix」_trimming_17mer)。スコア0がついている箇所がたくさんあるかと思いますが、先に頻度解析を実施するときに、ある程度長く一定以上のスコアがついていた部分だけが解析対象になっていたことを思い出してください(頻度解析の対象になっていなかった部分は、スコア0になる可能性があります)。
<その他のヒントと注意>
- multi FASTA形式のリードデータも処理できますが、以下の制約があります。この条件を満たしていれば、正常に計算されます(満たしていない場合はエラーが出ずに不正な処理結果が出力されます。ご注意ください)。multi FASTA形式のリードデータを処理する場合、プルダウンメニューより「multi FASTA」を選択してください。
- 1リードが1行に納まっているmulti FASTA形式であること
- 1リードの長さが255塩基以内
- 処理するとき、複数のイルミナファイル、multi FASTAファイルを指定できますが、一度に指定できるのはいずれかの形式のみです。
- ゲノム中のすべての場所がまんべんなく読めていれば良いのですが、イルミナではGC含量が高い部分が読まれにくいということがあるようです。極端にリードデータの厚みが薄いところは、本処理によってリードが除かれてしまうことがありますのでご注意ください。
- 17merに関しては最大31merまで変更することができます。また偶数値を指定するとそれを超えない奇数の値に自動的に変更されます。
- hash table sizeはデフォルトでは1000万ですが、ゲノムサイズが大きい場合はこの値を大きくすると速度が向上する場合があります。ただし、メモリを消費するので注意してください(1000万でおよそ200MB消費します)。
- max threadは、分岐する最大スレッド数を指定します。batch sizeは、何行をまとめて処理するかを指定します。お使いのMac、データのサイズなどによってこれらを変更するとパフォーマンスが向上することがあります(結果は変わりません)。
- 頻度データが準備できた後で、入力ファイル/ファイル形式を変更し、フィルタリングを実行することができます。
- 保存した頻度データは、読み込んで使用することができます。
- 単にクオリティスコアに依存して各リードの使用部分を決めるよりも、長い部分が利用可能になる場合があります。
missing k-mer探索機能
この機能では、まずリファレンスゲノムのDNA配列をk-merでhashした辞書を作成します。「k-merでhashした辞書」はリファレンス配列を、端から例えば21塩基とって辞書に項目として追加、また1塩基ずらして21塩基をとって辞書に項目として追加、という動作を繰り返して作成します。例えば7 Mbのゲノムからは、680万通りぐらいの21 merが作成されることになります(ゲノムの大きさより少し小さいのはゲノムの中に重複配列が存在するためです)。次いで、全部のリードについて、21塩基を、1塩基ずつずらしながらとりだして辞書をしらべ、辞書のその項目があればそれを削除します。このようにして辞書の中の項目を、消していくと、最後には「リファレンスゲノムには存在するが、どのリードにも登場しなかったk-mer」が残ることになります。
このようなk-merは、point mutation、insertion、deletionなどの変異箇所に関する情報を含んでいます。GenoFinisherは、全く見られなかったk-merのすべてと、それらをラフにアッセンブルしたものとの両方を出力します。
リードを抽出する機能
リードIDを指定して、(1)そのIDの配列、(2)そのペアとなるIDの配列、(3)これら両方を出力する機能があります。複数を同時に指定できます。多数を探す場合、grepなどを使って検索するよりも高速です。
任意のコンティグを形成するリードを詳細に解析する機能 (ContigDeepAnalysis)
別ベージにマニュアルがあります。