Finishingについて
ケース1: 454シーケンサー由来のシングルエンドデータのみがある場合は、newblerでアッセンブルしたのち、(1)「重複問題」をPCRにより解決、(2)またこのときバリエーション問題をPCR産物の配列決定によって解決、(3)「孤独末端問題」を総当たりPCRなどによって解決、することになります。ただしこの場合ホモポリマーサイトのクオリティが低いなどの問題が残ることになります。GenoFinisherは、行うべきPCRについての総合的な情報を出力します。またコンティグのつながりが明らかになった場合について、コンティグの連結を効率良く実施する機能を備えています。
ケース2: 454シーケンサー由来のペアエンドデータがある場合は、newblerでアッセンブルした時点で1つあるいはそれ以上の数のスキャフォールドが作られているはずです。この場合、(1)スキャフォールド内のgapの配列を明らかにし、また場合によっては(2)スキャフォールド間の配列を明らかにする必要があります。このケースでも、上のケースと同様にホモポリマーサイトのクオリティが低いなどの問題が残ることになります。GenoFinisherは、gapにはまることが推定されるコンティグルート及びDNA配列を出力し、また、gap部分を増幅するPCR primerを出力します。また、gapを埋めるのに利用できる可能性がある454リードを出力する機能があります。出力されたリードは再アッセンブルすることで、gap配列が得られる場合があります。gapに相当する配列を完全には得ることが出来ない場合もありますが、そのような場合であってもgapの配列についてある程度知ることができ、primerのデザインに便利です。またgap相当部分の配列を決定した場合、スキャフォールドの該当する部分に、決定した配列を挿入する機能があります。
ケース3:イルミナあるいはsolidのデータが得られていれば、ホモポリマーサイトについては修正できるものと思われます。修正には(1)これらデータをnewblerでアッセンブルする時の入力データに含めることで、ホモポリマーサイトの精度が良くなることを期待するやり方と、(2)ホモポリマーサイト以外についてはFinishできたゲノムについて、イルミナあるいはsolidのリードを利用して、ホモポリマーサイトを修正するやり方、が考えられます。(1)を採用した上で、さらに(2)を行って確認を行うのが良い(特にイルミナ/solidのデータ量があまり多くなく、ホモポリマーサイトの配列が454データに引っ張られて決まっていることが推定される場合)かと思います。GenoFinisherでは現在(2)についての機能の作成を検討中です。
ケース4: 454のシングルエンドデータと、イルミナのメイトペア-ペアエンドデータがある場合、両方をnewblerの入力とすれば、スキャフォールドデータを得ることができます。この場合も、ケース2と同様の処置に続いてケース3の処置が必要です。このようなケースでは、gapを埋めるのに利用できる可能性がある454リードを出力するだけでなく、gapを埋め得るイルミナリードも同時に出力します。
解決すべき問題の種類とその解決方法
細菌ゲノムを次世代シーケンサーで決定しようとしても、単に配列データをアッセンブルするだけでは完全決定にはいたらないのが現実です。Finishingするにあたり解決すべき問題は大きく分けて5つあります。これらの問題について本マニュアルでは以下の5つの言葉で呼びます。GenoFinisherはクオリティ問題以外については、それを解決するための機能を提供しています。
- 重複問題
- バリエーション問題
- 孤独末端問題
- ひげ問題
- クオリティ問題
重複問題
ゲノム中のリピート(重複配列)について考えます。例えば1 kbのある配列がゲノム中に2つある場合 (AおよびBとします。またAの直前の配列をa1、直後の配列をa2とします。同様にBについてはb1とb2とします)、1kbよりも短いリードデータをいくら解析しても、2つあるリピート領域それぞれについてその前後が、どれとつながっているかは明らかにできません(a1-A-a2なのかあるいはa1-A-b2なのか)。これを明らかにするには、a1、a2、b1、b2にそれぞれアニールするprimerを用いて、4通りのPCRを行い、どれとどれが繋がっているかを明らかにする必要があります。また後述するように、バリエーション問題がありますので、必要に応じてPCR産物の塩基配列決定を行う必要があります。
ペアエンドデータが得られている場合、基本的には重複問題は問題になりません(ゲノム中に例外的に長いリピートが存在すると、重複問題が残ることになります)が、リピートの存在に起因する「バリエーション問題」は解決されません。
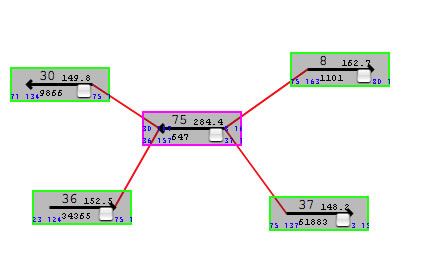
重複問題の例: コンティグ75 (647 bp)はリピート領域。コンティグ30がコンティグ75を中継してコンティグ8に繋がっているのか(30-75-8)、コンティグ37に繋がっているのかわからない(30-75-8)。30-75-8が正しいなら、36-75-37のはず。
バリエーション問題
newblerはコンティグを作るにあたり多数決を採用しているようです。リピート領域でない部分についてはそれで良いかと思うのですが、例えばゲノム中にある挿入配列が10コピーあり、そのうち1コピーに1塩基のバリエーションがあったらどうでしょうか?多数決(9対1)によってそのようなバリエーションは無視されてしまうようです。またこのような場合、その塩基に高いスコアがつけられていることにも注意すべきです。言い換えれば、重複度が高いリピート領域に関しては仮に全部の塩基に高いスコアがついていたとしても、配列に間違いがないことにはならないということです。また逆に、あるコンティグの重複度が2の場合で、その塩基すべてに高いスコアが付いている場合、リピートの配列にバリエーションはないと推測できます。
ペアデータがある場合では、アッセンブルするとスキャフォールドが作られます。スキャフォールドについては454Scaffolod.txtファイルに記述されていますが、スキャフォールドは基本的に、重複コンティグでないコンティグの序列であり、各コンティグの間にはGapが存在しています。454Scaffolds.fnaファイルを見るとこのGapはNの連続として表現されています。ここでFinishingに向けてすべきことは、このNNN・・・の部分をPCRで増幅して配列を決定し、NNN・・・の部分を置き換えることです。このGapの部分は、リードデータがないためにNNNになっていると思いがちですが、実際はそうではありません。コンティグ11から別のコンティグ12に行き着き得るルートを探索すると(GenoFinisherにその機能があります)、いくつかのルートに限定される場合がほとんどです。例えば、コンティグ11の後はコンティグ52でその次がコンティグ12であることがわかるということです。ではなぜあえてnewblerはNNNで出力するのかということに関しては、このようなコンティグ(ここではコンティグ52)はリピートコンティグであり、配列中に誤りがある可能性があるためである、と推測されます。
実際問題として、gapの隙間があまり長くない場合は、PCR増幅したあとに、両側からシーケンスを読むことでgapを決定することができます。たとえばサンガーシーケンサーでは安定して700bp程度の配列を得ることができるので、PCR増幅産物が1.4kb位であれば、両側からの一度のシーケンスによってgapの配列を決定することができます。しかし、例えば7 kbといった長いPCR増副産物の場合に、primer walkingによって読むのは時間がかかります。その意味で、あらかじめgapの配列がどんなものなのかあらかじめ類推できれば、複数のprimerを同時に発注することができ、結果として一度のシーケンスである程度長いgapを埋めることも可能となります。このようにあらかじめ各gap部分がどのような配列なのかを知ることができれば、効率良くgapクロージングができます。
また、gapを埋めることに関して、newblerはリードデータを余すところなく使えているかと言えばそうではありません。ペアデータとなるデータを利用することで(追加の実験なしで)gapを決定することが可能な場合があります。GenoFinisherの機能「ContigDeepAnalysis」では、重複コンティグがどのようなリードによりできているかを詳細に見ることができます。ContigDeepAnalysisでは、重複コンティグをつくるリードのうち、指定したコンティグと関連があるリードのみを表示することができます。この機能により、例えばcontig123がリピートコンティグであるとして、contig4とcontig5の間にあるcontig123について、配列に修正すべき箇所があるかどうかを知ることができます。
孤独末端問題
コンティグの片方の端、あるいは両方の端が、どのコンティグと繋がっているかわからない(454ContigGraph.txtに記述されていない)末端を孤独末端と呼びます。孤独末端はそのつながり先がわからないコンティグですので、事前の予想に基づいてPCRができる重複問題の場合と比べて、困難です。孤独末端のつながり相手を探すには2つの方法があります。
- 孤独末端間で総当たりPCRを行う。
- ゲノムを鋳型にして、シーケンス反応を行い、どのコンティグと繋がっているか調べる。
ゲノムを鋳型にしたシーケンス反応のプロトコールについては Environ. Microbiol. 12: 2539-2558 (2010) を参照してください。なお、ゲノム鋳型シーケンスは、抽出した全ゲノムDNAそのものを鋳型としてシーケンス反応に使う方法であり、一度PCRなどで増幅した後にその産物を鋳型にシーケンス反応を行う方法ではありません。
なお、454Finisherの作者があるゲノムを決定した経験では、孤独末端は11個(奇数!)しかなかったのですが、ゲノムを鋳型としたシーケンスによって、孤独末端の繋がり先がリピート領域であることが判明したことがありました)。なお、6個のGAPはいずれも大きな2次構造を取り得る配列のすぐそばに位置しており、コンティグの末端の配列数塩基の配列が誤っていました。このために、アッセンブラが別々のコンティグとして出力したものと思います。GAPのサイズは最大で10塩基程度でした。
ヒゲ問題
454で得る多数のリードの中には、本来ゲノムに含まれないと考えられるものが小数含まれるようです。これは、1) 細菌ポピュレーションの中に、ゲノム中に逆位などが起きた個体が小数存在していたため、あるいは、2) 454でのシーケンス反応中に生じたアーティファクトの塩基配列が読めてしまったため、であるかと思います。このようなもののうち、ある程度存在数が少ないものに関してはnewblerは無視するようになっているようですが、ある程度の本数に達すると、無視されなくなるようです。無視されるべきリードが、あらぬコンティグ同士をつないでいると解釈されてしまった場合、この繋がりは比較的薄い(つなぐリード数が少ない)ものとなります。GenoFinisherでは、コンティグ間をつなぐリードに比例した太さの赤線が、コンティグ間に引かれます。そのためこのような薄い繋がりは、薄い線でつながれることになります。このような意味のない薄い繋がりを、本マニュアルでは「ヒゲ」と呼びます。薄い繋がりについては、無視するような設定ができます。
ペアデータがあり、十分少ない数のスキャフォールドが得られている場合はヒゲ問題に悩まされることはあまりないかと思います。
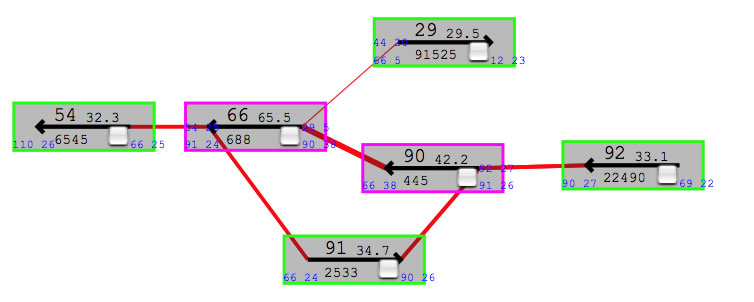
コンティグ66とコンティグ29をつないでいるのがヒゲ。このヒゲのせいで、コンティグ66とコンティグ90は2つのコンティグとしてnewblerによって出力されていると思われる。赤線の太さが、つなぐリードの数に比例していることに注意。またコンティグのカバレージはコンティグ番号の右側に示されている(コンティグ54のカバレージは32.3).
クオリティ問題
良く知られている通り、454シーケンサーはホモポリマーサイトを苦手とします。要するに同じ塩基が連続している場合に、エラーが多いということです(ゲノムが既知の株について、454のリードを得て付属のGS mapperで解析すると、どれ位間違えたかを知ることができます)。これに関しては、何らかの別のシーケンスデータを追加しない限り、解決しません。