
SeqView is a software to help you understand how DNA and amino acid sequences are related. SeqView is available from App Store for free.
It often happens that you have several DNA sequences, and you want to know how they are related to each other. The main concept of SeqView is that it would be nice if you can place and move DNA sequences on a 2D view panel and compare them to each other.
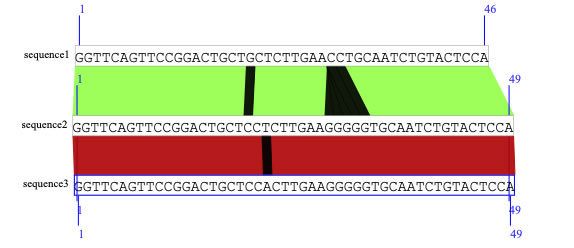
As you can see below, DNA sequences are compared, and similarities are shown by colored rectangles, while mismatches are shown by black lines.
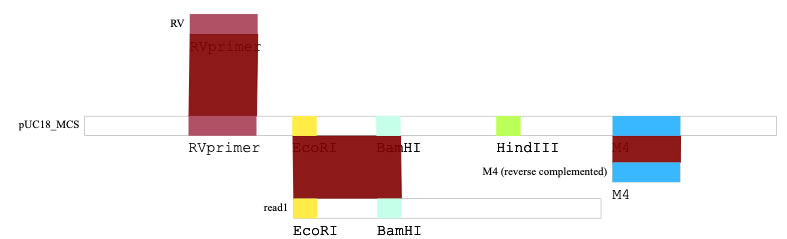
You can color DNA sequences. The image below shows the multi-cloning-site of pUC18, surrounded by M4 and RV primer annealing sites.

DNAs are also shown by bars.
Inputing sequences
The following three DNA sequences are in the FASTA format.
>sequence1
GGTTCAGTTCCGGACTGCTGCTCTTGAACCTGCAATCTGTACTCCACCCAAGAAGGCGAACAGCCGCTTACAGGGCAAAATGACGGATTCGGCAG
>sequence2
GGTTCAGTTCCGGACTGCTCCTCTTGAAGGGGGTGCAATCTGTACTCCACCCAAGAAGGCGAACAGCCGCTTACAGGGCAAAATGACGGATTGGGCA
>sequence3
GGTTCAGTTCCGGACTGCTCCACTTGAAGGGGGTGCAATCTGTACTCCACCCAAGAAGGCGAACAGCCGCTTACAGGGCAAAATGACGGATTGGGCA
Copy and paste the three DNA sequences (red characters altogether) into the text field in the "adding sequences box", then click the add button. Now DNA sequences are displayed in the main view.
Selecting sequences
You can click on a sequence to select, and the selected DNA sequence will have blue border lines. If you click the next one, the selected one is deselected, and the new one is selected. Click and drag to move the sequence around.
You can also select DNA sequences by clicking at an open space and drag to the right to select fully enclosed sequences. Drag to the left to select sequences that intersected with the drag rectangle.
Multiple selections are also possible by clicking DNA sequences while pressing the shift key.
To reverse-complement the sequence, select "invert panel" from the context menu.
Comparing sequences
Now let's compare two sequences by blastn. To do this, select two sequences (assuming sequences 1 and 2), and check if the two sequences have blue borders. Click the run button in the "blast comparison" box. Now you see a colored rectangle, which indicates detected similarity, and the black lines show nucleotide mismatch.

The color indicates the similarity between the two sequences.
To check the color scale, press "command+ ," to open the preference window.
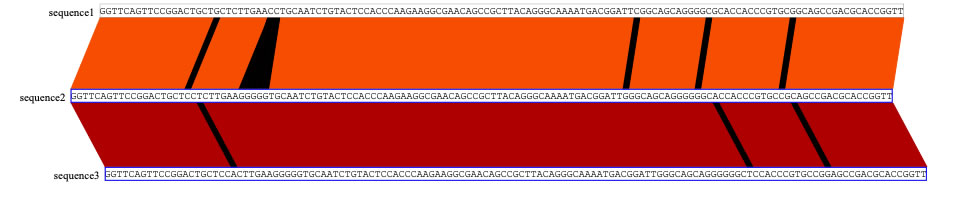
Now select the other pair (sequences 2 and 3) to run the blastn.
Finding and coloring sequences
Let's use the "find and mark" function. To the text filed in the "find and mark box", type "red ACCC" and press the find and mark button. Now, the sequences of ACCC and also GGGT (complementary to ACCC) are colored in red. Here, the alpha value (opacity) is set to 50%.

To show ACCC only, uncheck the "both strand" check box. The colored sequences now also have the color name 'red' just beneath the found sequences. To not show the color-key (here 'red'), uncheck the "color key" check box.
You can add "EcoRI GAATTC" in the new line of the text filed. Now the color-key is "EcoRI." To edit the association between a color-key and a color, open the color setting window (command + K), and click a color well next to the color-key of interest, and pick a color as you want.
If you use a color-key that is not set in the color setting window, a random color is created and associated with the color key. If you omit to set a color key, the sequence itself is used as a color key.
You can set to find multiple sequences simultaneously, and in case the text field is too small, use the enlarge button.
Selecting a DNA region
By double-clicking on the DNA sequence, you can start selecting a DNA region. For selection, click-and-drag the sequence, and adjust the selection by using the left and right arrow keys. As you select, the location within the DNA sequence, the length of the selected DNA tract, and a Tm value are shown.

You can give a name to a specific DNA region. To do so, set a name candidate in the "name candidate" box, for example, "Lac promoter". Then select the relevant region, and right-click the sequence and from the context menu, select "register the selected region as XXXX", and here XXXX is the candidate name. In case the candidate name is not unique among the names you have already given, the menu is grayed out.
Once you register a region, you can scroll to the region from the context menu. The registration can be erased from the context menu.
Two displaying modes; "Sequence mode" and "bar mode"
There are two appearance modes; sequence mode and bar mode. In the bar mode, sequences of A, C, G, and T are displayed, and in the bar mode, rectangles (bars) are displayed. In the bar mode, you can set a drawing ratio in points/kb, i.e., if 0.1 is set DNA sequence of 1000 bp is displayed as a bar with a width of 100 pt. The bar mode was so designed that large DNA sequences could be displayed.
Now let's compare contigs assembled from Illumina reads with a large genomic sequence created by PacBio in the bar mode. The drawing ratio is now 0.3. Now there are 29 contigs, and to compare those with a single sequence, select the 26 contigs as described above, and command+click the later sequence to set it as a target. The target sequence's borders are shown in red. By pressing the run button, contigs are blasted against the target sequence. The contigs are automatically aligned to the target.
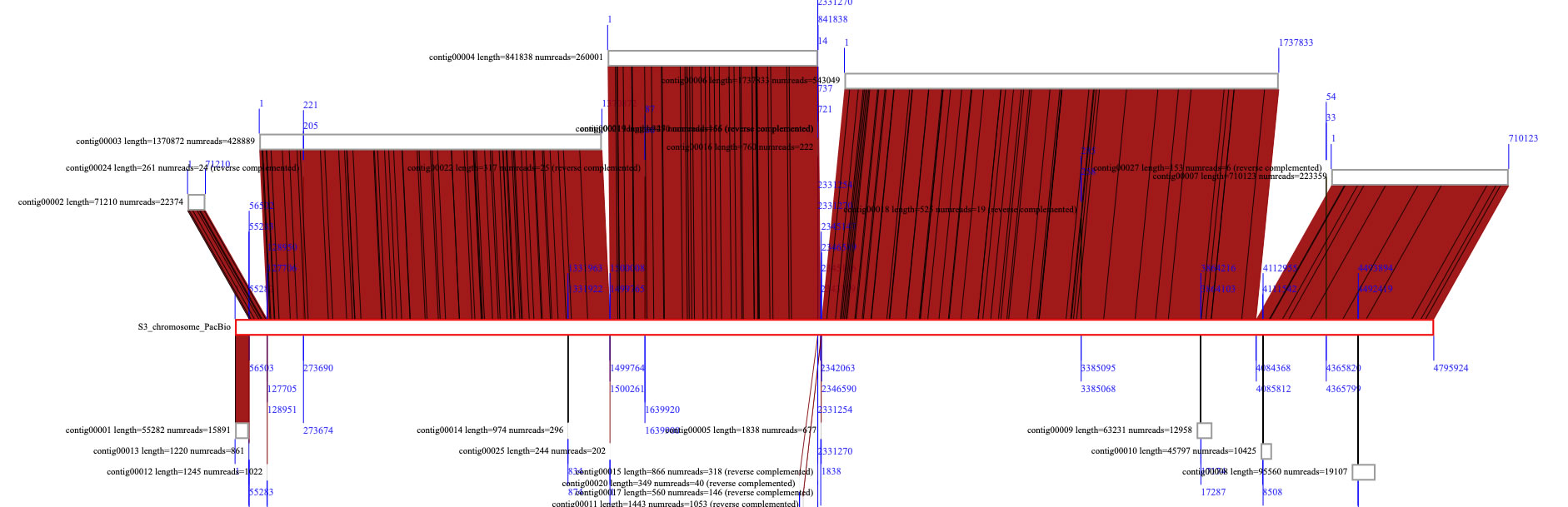
To zoom up to a specific region, right-click the view panel and select "zoom up here in sequence mode" or select the icon for zooming in, and drag over a region where you want to zoom.
Use the left and right arrow keys to scroll. The scrolling size is calculated from the value set in the "scrolling by arrow keys" box. If the value is 100, the view is scrolled by 100 nucleotides.
Finding a DNA sequence
Use the pulldown selector to find a sequence by its name. Upon selecting a sequence name, the view is scrolled to show the sequence, and the sequnce blinks twice.
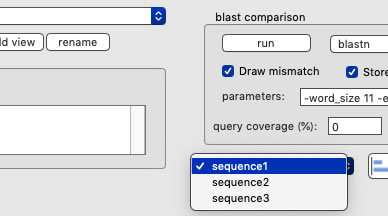
Creating a new view
Upon launching, the name of the view is "View1", which is marked at the left-top corner of the view. To rename, set a new name and press the "rename" button. To add a new view, set a new name, and press the "add view" button. Use the pulldown menu to change a view to show.
Saving the image in a PDF file
From the file menu, choose "save PDF image" or "save all PDF images", to save the drawings into PDF files.
Blast parameter settings
Refer to the blast+ document for blast parameters. In SeqView, there are four threshold values to avoid drawing non-relevant similarities. You can set a query coverage threshold, length threshold, and identity threshold. Similarities not exceeding the threshold values are ignored. However, note that setting a value of 0 means not applying that threshold.
The last one is the "hits to store" threshold. If the value is 3, for each query, data for the top three HSP are stored for drawing.
Where all selected sequences have blue borders, all sequences are used to create a blastdb, and all of the sequences are blasted against the database (self-to-self hit is ignored). Where one or more sequences have red borders, the sequences with red borders are used to create a blastdb, and sequences with blue borders are blasted against the database.
Amino acid sequences
You can also handle amino acid sequences.I would appreciate your sending me bug reports.
Yoshiyuki Ohtsubo Ph.D..
yoshiyuki.ohtsubo.a6[at]tohoku.ac.jp
I would appreciate your considerations of the donation to my laboratory. Now I am trying to develop a rapid detection system for the new coronavirus, SARS-CoV-2. My challenges are to develop a means to detect SARS-CoV-2 within two minutes. I have already obtained a pool of DNA aptamers that binds to the receptor-binding domain of the spike protein (S-RBD). However, I have been facing difficulties in getting found for further development. The donation will be used for the development as well as to support software engineering, and to conduct research and educational activities in my laboratory.
Please fill the donation application form and e-mail it to our accounting office: lif-kaik"at"grp.tohoku.ac.jp
 大学改革の方針転換を。「REUP提案」をお読みください。
大学改革の方針転換を。「REUP提案」をお読みください。
 入試や就活で「推し研」を問いませんか?
入試や就活で「推し研」を問いませんか?
 本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
 本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
 QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
 TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
 ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
 GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
 GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
 DNA配列の相補配列を計算する関数を含むエクセルシートです。
DNA配列の相補配列を計算する関数を含むエクセルシートです。
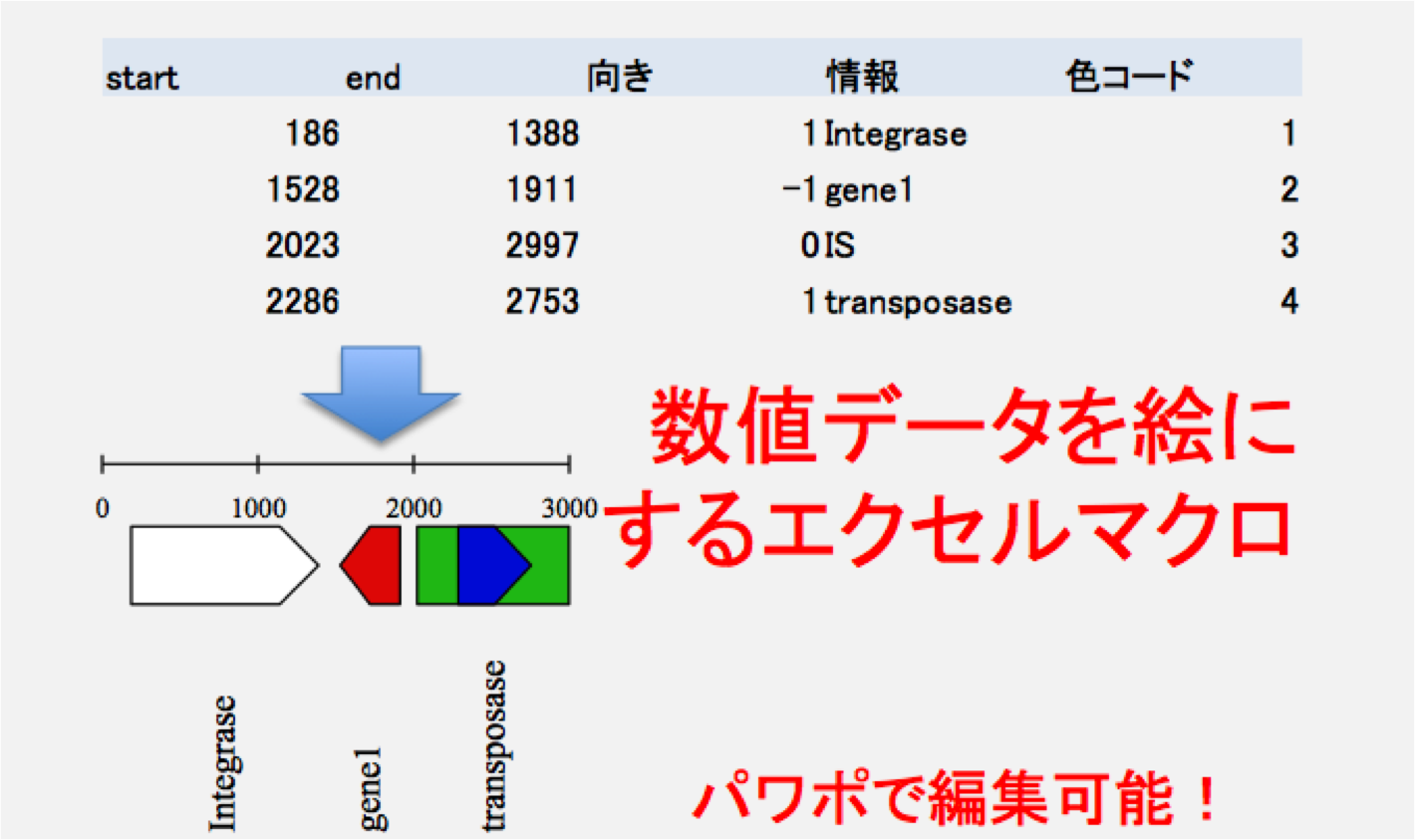 数値データを絵に変換するマクロを含むエクセルシートです。
数値データを絵に変換するマクロを含むエクセルシートです。
 数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
 大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
 DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
 例の処理を簡単に済ませるあのツールです。
例の処理を簡単に済ませるあのツールです。
 iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
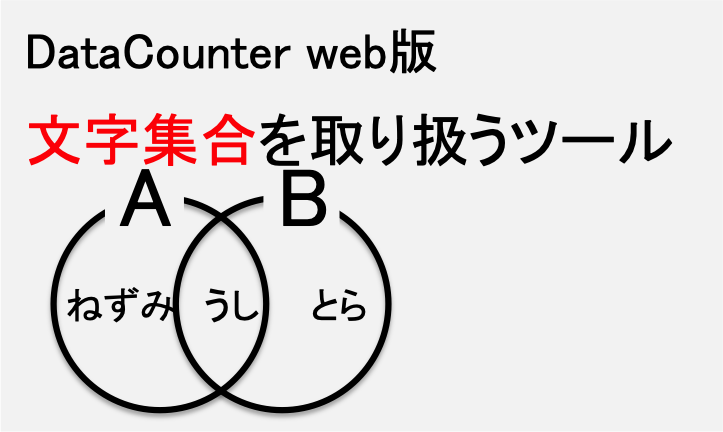 文字列集合を取り扱える便利ツールです。
文字列集合を取り扱える便利ツールです。
 GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
 文字列を取り扱える便利ツールです。
文字列を取り扱える便利ツールです。
 作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。
作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。
作者プロフィール: 環境細菌の研究を進める一方で、様々なソフトウエアを作成、公開している。