TraceViewerForMolecularBiologyは、キャピラリーシーケンサーで取得したデータを解析するためのソフトウエアです。App Storeから配信中です。
キャピラリーシーケンサーは高性能であるにも関わらず、ほぼDNAの配列解析にのみ用いられてきましたが、従来変性ポリアクリルアミドゲル電気泳動で行なっていたような分子生物学的DNA解析にも利用することができます。
ただしキャピラリーごと、泳動ごとに若干泳動度が異なるため、データを相互に比較するには泳動度を補正する仕組みが必要です。蛍光ラベルされたサンプルに、別の蛍光ラベルサイズスタンダードを混ぜて解析すれば、サイズスタンダードを使って泳動度を補正することができます。TraceViewerForMolecularBiologyは泳動度を補正する仕組みや、その他作図用機能、ピーク面積の出力機能など多数の機能を備えています。
シーケンサーをDNAの配列決定だけでなく、DNAの分子生物学的解析にも使おうではないか、というのが本アプリの名前の由来です。
本アプリは作者が自分の解析のために作成したアプリですが、多分便利だと思ってもらえるように思いますので、広く公開することにしました。
変性ポリアクリルアミドゲル電気泳動はかなり大変な実験です。ゲル作りに5時間、泳動に12時間、検出に12時間以上かけるのが普通です。解析できるサンプルの数は48サンプル程度です。また確実にうまくいく実験手法ではありません。ゲルが大きくて薄いために取り扱いが難しく破けてしまう場合もあります。また望ましくない汚れが大事な部分を隠してしまったりします。そのような時、サンプル調製からやり直しになる場合もあります。サンプルが濃いほど良いデータが取れるのでサンプルは1度の解析で使い切ってしまいがちです。熟練した場合でもおそらく2日に1度データが取れれば良い方であると思います。
一方キャピラリーシーケンサーではどうでしょうか?まずゲルを調製する必要がありません。泳動時間25分程度で200塩基ぐらいまでのデータが取れます。時間をかければ1200塩基のデータも取れます。解析できるサンプル数はシーケンサーにもよりますが4から16程度でしょうか?サンプルをrunしてデータを得るまで1時間以内でしょう。結果を見て次の実験をする、というサイクルを例えば1日に4サイクル程度行うことも可能でした。感度も良く単一のDNAが1 fmolあればシグナルが振り切れるほどです。ラジオアイソトープを使った実験では1 laneあたり10 fmol程度でしょうか?それよりもずっと少なくて済みます。しかもサンプルのごく一部がキャピラリーにインジェクションされるだけですのでサンプルは繰り返し分析できます。つまりシーケンサーの都合でrunがうまくいかなかった場合でもサンプルが失われず、もう一度解析ができます。しかもシーケンサーだけに解像度が抜群に良く、1塩基の解像度で解析ができます(解像度が最も良く得られるところでは、1塩基以下の解像度が得られます)。
上記のようにキャピラリーシーケンサーは極めて優れた解析能力を持っています。これをDNA配列解析装置としてだけ用いるのは非常にもったいないことです。
データ形式
TraceViewerForMolecularBiologyは、ABIFフォーマットのデータを読み込んで解析するためのソフトウエアです。したがって本アプリを利用して解析を行うには、ABIFフォーマットの出力が可能なシーケンサーを使用する必要があります。ABIのシーケンサー(日立製)ではABIF形式のデータを出力します。受託解析サービスではおそらくABIシーケンサーを使用していますので、ABIF形式データが入手可能だと思います(ご確認ください)。
蛍光ラベル
解析対象DNAは蛍光ラベルされている必要があります。また蛍光ラベルされたサイズスタンダードが必要です。解析対象につける蛍光基とサイズスタンダードの蛍光基は、蛍光スペクトルが十分に離れているのが望ましいです。作者は、解析対象をFAMでラベルし、サイズスタンダードとしてはLIZラベルマーカーを使用しています。LIZラベルサイズマーカーとしてはサーモフィッシャーサイエンティフィック社の500LIZ、あるいは1200LIZサイズスタンダードを使用しています。
5'-FAMラベルしたオリゴDNAは、ユーロフィンジェノミクスなど多くのDNA合成サービスを利用して購入できます。
解析用DNAの調製
FAMラベルされたオリゴDNAを用いてPCRをすれば2本鎖DNAを調整できます。短いDNAは、PCRの代わりに2本のDNAをアニーリングして調製します。作者の場合、PCRで増幅する場合には、増幅後、PCR反応溶液にExonuclease Iを加えて一本鎖DNAを分解し、ついでサンプルをポリアクリルアミドゲル(スーパーセップ エース)で泳動してバンドを切り出し、クラッシュ法でDNAを回収します。クラッシュ法にはバイオマッシャーIIを使用しています。PCRに用いたprimerの5から20 %程度が得られます。
サンプル調製
LIZサイズスタンダードは、あらかじめHiDi formamideに加えておきます。マニュアルでは1 mlのHiDi formamideに20 µl加えることになっていたと思いますが、節約のため1 mlに2 µlを加えます。シグナルが弱く実用に耐えなければ加える量を増やします。サイズスタンダードを加えたHiDi formamideをサンプルプレートに12 µlずつ分注しておき、ここに解析したいサンプルを加えます。加えるサンプルの量は0.1から2 µl程度です。加えるサンプルの塩濃度が高いとシグナルが弱くなります。必要があれば、エタノール沈殿、カラム精製などを行なってからサンプルを加えます。ゲノム微生物学会のニュースレター19号に、DNA精製用のミニカラムの作成方法を紹介した記事があります。
単一のサイズのDNA分子しかなく、また十分に精製されている場合、加えるDNA量を1 wellあたり1 fmol以下となるようにします。かなり少ない量ですが、この量でシグナルが飽和すると思います。反応buffer由来の塩が混入する場合では解析対象DNA量は多くしても大丈夫ですが、マーカーシグナルが弱くなってしまいます。
サンプルを熱処理する必要はありません。
分解産物由来のシグナル
蛍光基でラベルされたDNAを分解すると、分解されるに連れて泳動速度が速くなっていきます。FAMラベルされた20塩基のDNAの3'が1塩基短くなったDNAは、より早く検出器に到達します。ところがある程度短くなると(おそらく蛍光基+2塩基)突然泳動速度が遅くなって、75ヌクレオチド程度の位置にシグナルがでるようになります。蛍光基の大きさや電荷によってこのようになるのだと思います。
サイズの正確性
DNAの泳動速度は、DNAの配列によって異なります。つまり100ヌクレオチドの断片2つを混ぜて解析した場合、これらは2つの異なるピークとして検出されます。つまり、例えばLIZ500マーカーの100ヌクレオチドと同じ位置にピークが検出されたからといって、その断片の長さが100ヌクレオチドであるということには必ずしもなりません。サイズマーカーはおおよそのサイズを知るための手段にしかならない(正確なサイズを知ることはできない)ということに注意してください。
配列によって泳動度が異なるということは、例えば、3'末端がA、C、G、Tである点が異なる以外はお互いに同じ配列である4つのDNAの泳動度が微妙に異なるということからも確認できます。3'端がGあるいはAであるものは、泳動速度がCあるいはTであるものよりも遅いです。
PCRの時にそれぞれのpiremrを別々の蛍光基でラベルし、これを解析すると、2つの蛍光基は異なる場所にピークとして観察されます。これは2つのストランドの配列が異なる(お互いに相補的であってもAAAAとTTTTは異なります)ためであると解釈できます。
つまりサイズを正確に知りたかったら、配列が全く同じであるサイズコントロールを用意して比べる必要があります。LIZ500などのサイズスタンダードは泳動速度のキャリブレーションにのみ用いるのが良いと思います。
1. 広く用いられている逆転写酵素に、強いtailing活性があることを見出しました。従来2本鎖DNAの3'末端に単一のAを付加する活性をTaqポリメラーゼが有していることが知られていましたが、C、G、Tをある程度の限定された数を付加可能な活性は知られていませんでした(ターミナルデオキシヌクレオチジルトランスフェラーゼTdTは際限なく付加してしまいます)。この論文では、ACGTのいずれをも付加する活性を、マウス白血病ウィルス由来の逆転写酵素(MMLV-RT)が有していることを報告しました。MMLV-RT由来の逆転写酵素は、リバトラエース(TOYOBO)、スーパースクリプト(Thermo Fisher Scientific)、SMARTScribe(TAKARA)などとして販売されています。
Ohtsubo Y, Nagata Y, Tsuda M. 2017. Efficient N-tailing of blunt DNA ends by Moloney murine leukemia virus reverse transcriptase. Scientific Reports 7, 417692. 上記の活性を促進する化合物を見出しました。例えばGの付加反応を200 mMのデオキシシチジンが強く促進し、4個程度のGを付加することができます。促進化合物によりCやTも3個数程度突出させることができます。
Ohtsubo Y, Nagata Y, Tsuda M. 2017. Compounds that enhance the tailing activity of Moloney murine leukemia virus reverse transcriptase. Scientific Reports 7(1):65203. 2本鎖DNAの3'端から突出させたGGGGに、3'末端にCCCCを有する一本鎖DNAを作用させ、MMLV-RTによってその相補鎖合成を伴って効率良く取り込ませることができることを示しました。
Ohtsubo Y, Sasaki H, Nagata Y, Tsuda M. 2018. Optimization of single strand DNA incorporation reaction by Moloney murine leukaemia virus reverse transcriptase. DNA Research 25(5):477-487.4. 超音波で断片化したDNAの末端構造について知見を得ました。従来物理的に断片化したDNAの5'末端にはリン酸基、3'末端には水基があるとされてきました。TraceViewerを用いた解析により、超音波で破砕したDNAの3'端のおよそ半数にはリン酸基がついていること、30%程度が水酸基であること、残り20%は未知の構造となっていることを見出しました。またこれらを踏まえて、効率の良い末端修復方法SB-reparing法を開発しました。市販の末端修復キットには、5'端をリン酸化するためと称してT4 polynucleotide kinaseが含められているものがありますが、実は、T4 polynucleotide kinaseの3'リン酸を除去する活性が、末端修復に重要な役割をしていることを指摘しました。FAMはT4 polynucleotide kinaseによってリン酸化されて泳動速度が変化するので注意が必要です。
Ohtsubo Y, Sakai K, Nagata Y, Tsuda M. Properties and efficient scrap-and-build repairing of mechanically sheared 3' DNA ends. Commun Biol. 2019 Nov 8;2:409. doi: 10.1038/s42003-019-0660-7.5. 鉄硫黄クラスターをeffectorとする転写因子IscRのDNA結合部位をDNase Iフットプリンティング解析により明らかにしました。また、in vitro transcription産物に、5' FAMラベルされた逆転写primerをアsニーリングして、転写産物を検出することで、IscRが転写活性化能を示すことを確認しました。
Nonoyama S, Kishida K, Sakai K, Nagata Y, Ohtsubo Y, Tsuda M. 2020. A transcriptional regulator, IscR, of Burkholderia multivorans acts as both repressor and activator for transcription of iron-sulfur cluster-biosynthetic isc operon. Res Microbiol. 171(8):319-330.6. プラスミドの水平伝達において、接合伝達開始部位(oriT)中のnickサイトを同定しました。oriT含有DNA断片の片方の5'端をFAMラベルし、これに精製したリラクサーゼタンパク質を混ぜてnickを導入させ、生じる断片長を調べました。
Kishida K, Inoue K, Ohtsubo Y, Nagata Y, Tsuda M. 2016. Host range of the conjugative transfer system of IncP-9 naphthalene-catabolic plasmid NAH7 and characterization of its oriT region and relaxase. Appl Environ Microbiol 83:1e02359-16
サンプル波形データ
テスト用データ をダウンロードしてください。波形ファイル9つ分です。片側の5'末端がFAMラベルされた70 bpのDNAを基質として、dATP、dCTP、dGTP、あるいはdTTPとMMLV-RTを反応させた時のデータです。10 µlの反応系で15 minあるいは30 min反応後に、0.5 µlをサンプリングして解析した結果です。
波形データの読み込み
FileメニューのLoad trace fileから波形データを読み込みます。データはABIF形式である必要があります。ファイルがABIF形式であるかどうか調べるには、ターミナルでhead -c 4 に続けてファイルパスを指定します。これでABIFと表示されたらABIF形式です(ABIF形式のファイルでは、ファイル先頭の4文字がABIFであることになっています)。ソフトウエアによってはABIF形式のデータファイルを勝手に別の形式に変更してしまうものもあるようですので気をつけてください。
ファイルを読み込むとデータテーブルにデータが追加されます。このデータテーブルはコマンド+Dで前面に表示されます。ファイル中のデータは読み込まれるだけで、アプリはデータファイルに変更を加えません。
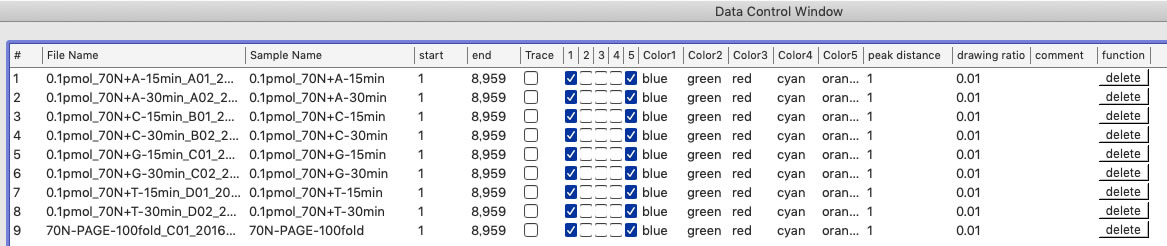
startとendは、キャリブレーションバンドの位置。初期値は1(start)と最後のデータ番号(end)。Traceチェックボックスは、そのデータをTrace Viewで設定するかどうか。1から5のチェックボックスはそのチャンネルデータを表示するかどうか。color1から5はチャンネル1から5のデータを表示する時の色を指定するカラーキー。drawing ratioは縦軸方向の表示倍率。シグナル値が5000でratioが0.01なら、ベースラインから上50ポイントのところに描画される。comment欄の文字列はTrace Viewパネル右側に表示される。
データは、コントルール+上矢印、あるいは、コントロール+下矢印で順番を変えられます。また「Sample Name」カラムのヘッダー部分をクリックすると、Sample名でソートされます。Sample名にアンダーバーが含まれている場合、アンダーバーを区切り文字として、第1フィールド、第2フィールド....でソートされます。サンプル名に気をつけておくと、ソートが楽になります。
このテーブルで、データの表示、非表示を切り替えたり、線の色を変えたり、キャリブレーション用マーカーの位置を設定したり、縦方向の拡大倍率を変更したり、データにコメントをつけたりできます。 色はカラーキーで設定します。カラーキーと実際の色の関係はEditメニュー中の、Show Color Panelで開くwindow中で確認、設定できます。チェックボックスのカラムについては、カラムヘッダーをクリックすると設定を反転できます。peak distanceで数値を設定すると、Trace Viewの画面で、キャリブレーションピーク間に薄い縦線が入ります。peka distanceに25を設定すれば、24本の縦線が均等に入ります。
5個のチャンネルのうち1がFAM、6がLIZのチャンネルです(Dye Set G5を使用の場合)。
キャリブレーションバンドの選択
コマンド+Rで「Raw Data View」を開いてください。下にレーン番号がありますが、これはデータテーブルの左端のカラムの番号と対応しています。
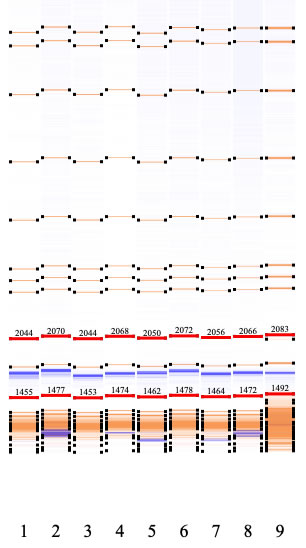
青はFAMの、オレンジはLIZのシグナル
キャリブレーションに使用するバンドをクリックして選択してください。黒いドットが両端にあるのは自動認識されたバンドです。1レーンあたり2つ選択する必要があります。選択されたバンドには赤い線でマークされます。複数のバンドを同時に選択したいときはオプションを押しながらドラッグしてください(範囲に含まれる一番上と一番下のバンドが選択されます)。
選択されたキャリブレーションバンド位置(選択されたバンドの上に表示されています)は、データテーブルに表示されます。データテーブルの値は、手入力で変更することもできます。キャリブレーションに使用されるのは、データテーブルにある値です。なお、Raw Data View中でシフトを押しながらレーンをクリック+ドラッグするとドラッグ位置が表示されます。
波形データの解析
コマンド+TでTrace Viewウィンドウを前面にします。このウィンドウでは、データファイル1つあたり1つのパネル上にデータが表示されます。
キャリブレーションピークの確認
calibratorsにチェックを入れて、適切にキャリブレーションピークが選択されているか確認してください。シフトを押しながらピークをクリック+ドラッグすると、その位置が表示されます。必要に応じてデータテーブル中の座標を手で修正してください。
見た目の変更
ピークの追加
コンテクストメニューから、「add peak」でピークを追加できます。
ピークの選択と編集
enable and show selectionにチェックを入れてください。これでピークをクリックで選択できるようになります。
ピークをダブルクリックするとピーク編集モードに入ります。編集モードでは、左右の境界線(緑)と、ベースライン(赤)、ピーク中央(青)が表示されます。左右境界線はつまんで動かせます。またベースラインはつまんで動かせる他、上下矢印キーで動かせます。編集モードから抜けるには、ピークから十分離れた場所をクリックしてください。
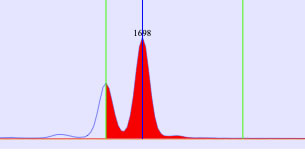
ピーク編集中の様子
ピークの面積合わせ
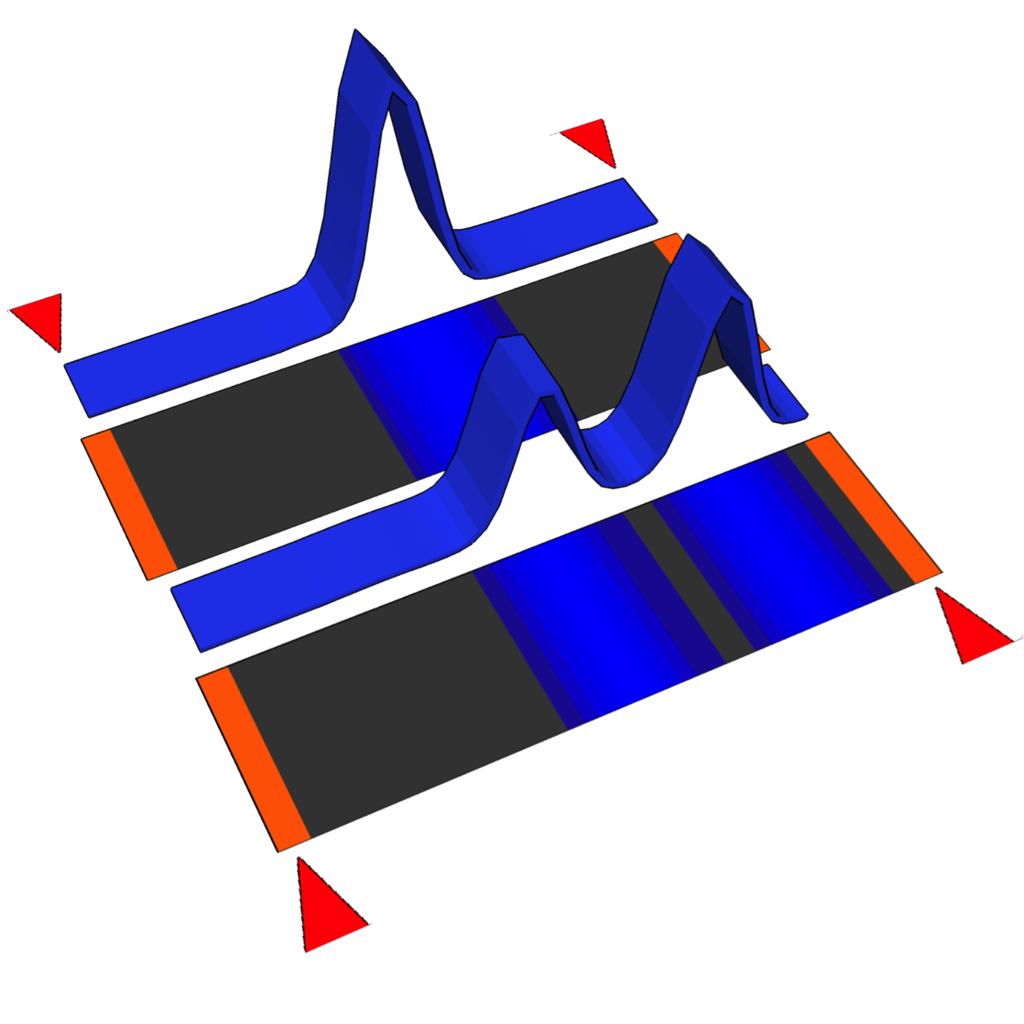
ピークが選ばれた状態で「even out peaks」ボタンを押すと、選択されたピークの総面積が全パネルで同じになるように、拡大倍率が調整されます(少なくとも1つ以上ピークが選択されている最も上にあるパネルの、選択ピークの面積の総和と同じになります)。下図はピークを選択したところです。
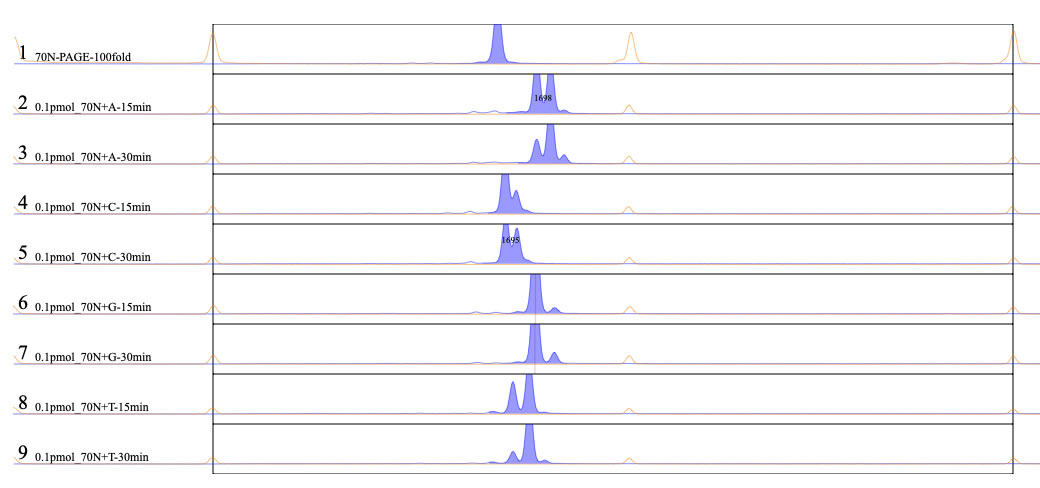
青はFAMの、オレンジはLIZのシグナル。黒枠はキャリブレーションピークの位置を示している。
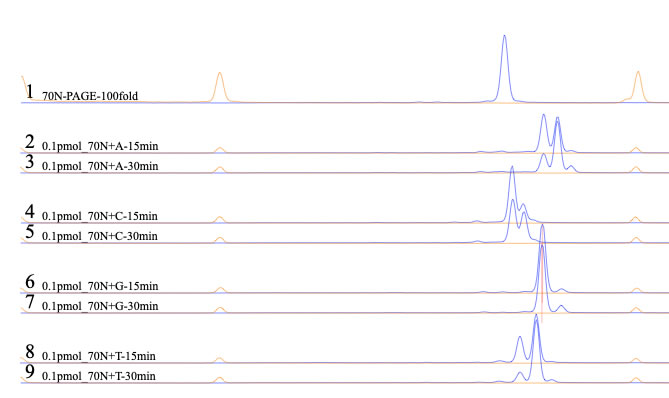
選択されたピークの面積を揃えて表示している。1は反応前。sample 7ではシグナルが飽和しているとされたところが僅かにある。パネルの高さは80ポイント、panel間のgapとして-30,-60を指定してある。オレンジの線を非表示に、青い線を黒に変更してPDFで書き出せば、論文用の原図が簡単に作成できる。
面積の出力
「copy selected peak data to clipboard」ボタンで、ペーストボードにデータがコピーされます。表計算シートなどにペーストしてください。
PDF出力
「PDF」ボタンからデータを出力してください。
PDFマニュアルです。
始めるのには少々困難を感じることがあるかもしれません。サポートしますので、PIの方よりメールにてご相談ください。科研費などプロジェクト分担者などへの参加依頼も歓迎します。
本ソフトウエアは無保証です。バグを見つけましたらご報告いただけますと助かります。
作者: 東北大学大学院生命科学研究科 准教授 大坪嘉行
yoshiyuki.ohtsubo.a6[アット]tohoku.ac.jp
本ソフトウエアを利用した成果を公表する場合、以下の論文を引用してください。
Ohtsubo Y, Nagata Y, Tsuda M. 2017. Efficient N-tailing of blunt DNA ends by Moloney murine leukemia virus reverse transcriptase. Scientific Reports 7, 41769
作者は現在、新型コロナウィルスを2分程度で検出可能な迅速検出法の構築を目指しています。これまでに新型コロナウィルスに結合するDNAアプタマーを取得し、これを複数装荷した分子を作製しているところです。このような分子はコロナルウィルスが存在すると凝集体を形成させ、これを容易に検出できるものと期待しています。これまで複数の研究助成などに応募してきましたが、残念ながら研究費の獲得に至っておりません。この研究を続けるため、私の研究室へのご寄附をご検討いただければ幸いです。
寄附金は、新型コロナウィルス関連の研究ならびにソフトウエア開発などの研究教育を目的とした活動に有効に利用させていただきます(大学によって通常の研究費と同等に管理されます)。寄附金についての本学での取り扱い、税制上の優遇措置などについてはこちらをご覧ください。
ご寄附頂ける場合は、専用の様式にご記入いただき、東北大学生命科学研究科会計係までメールにてお送りください。ご検討のほど、よろしくお願いいたします。
様式はこちらからダウンロードしてください。メールアドレスはlif-kaik[at]grp.tohoku.ac.jpです。
 大学改革の方針転換を。「REUP提案」をお読みください。
大学改革の方針転換を。「REUP提案」をお読みください。
 入試や就活で「推し研」を問いませんか?
入試や就活で「推し研」を問いませんか?
 本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
 本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
 QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
 TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
 ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
 GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
 GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
 DNA配列の相補配列を計算する関数を含むエクセルシートです。
DNA配列の相補配列を計算する関数を含むエクセルシートです。
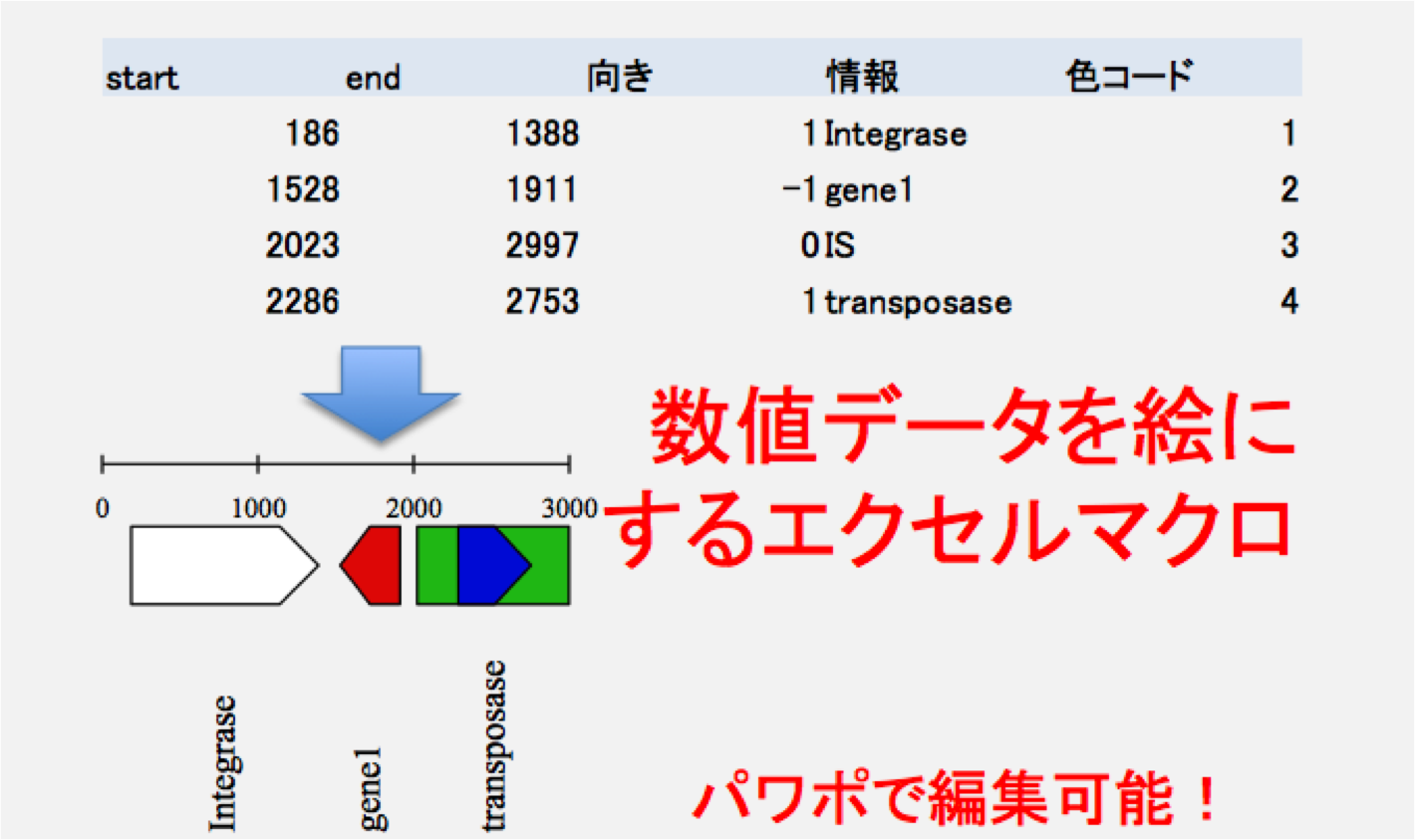 数値データを絵に変換するマクロを含むエクセルシートです。
数値データを絵に変換するマクロを含むエクセルシートです。
 数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
 大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
 DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
 例の処理を簡単に済ませるあのツールです。
例の処理を簡単に済ませるあのツールです。
 iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
 文字列集合を取り扱える便利ツールです。
文字列集合を取り扱える便利ツールです。
 GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
 文字列を取り扱える便利ツールです。
文字列を取り扱える便利ツールです。
 作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。
作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。
作者プロフィール: 環境細菌の研究を進める一方で、様々なソフトウエアを作成、公開している。