
ShortReadManagerは、NGSリードを処理するためのアプリです。
App Storeからの配信を開始しました。
TweetNGSリードには、エラーが含まれていますが、これを除去することができます。クオリティスコアによる除去とは異なります。
NGSリードのエラー部位を除去すると、その後の解析が速くなります。フィニッシングであれば、コンティグを構成するリードのアラインメントを見たときに、不一致箇所がなくなります。
処理しておけば、その後の解析で、ほんの1本とか2本とかのエラーを含むリードに惑わされることがなくなります。
たくさんのリードについて、まず登場するk-mer全てについて何回登場しているか数え上げます。ここでkは整数を意味しており、例えば21-merなどです。
得られたデータについて、横軸にk-merの登場回数、縦軸にk-merの数をプロットすると例えば下図のようになります。
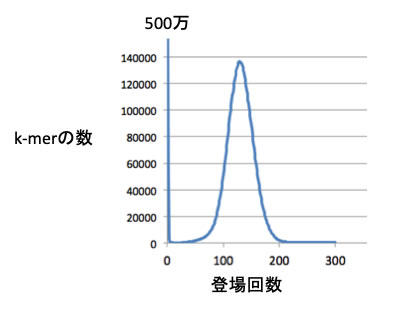
このグラフからは100回登場したk-merが4万通り、120回登場したk-merが14万通りあったことがわかります。注目して欲しいのは、1回しか登場していないk-merが500万通りもあるということです。
この500万通りもある1回しか登場しないk-merは、偶発的な何か、例えば読み取りエラーとか、アダプターとのジャンクション部分に由来することが考えられます。
続けて、全部のリードについて、登場頻度の下限値をセットして、それよりも低い部分を除去します。例えば2回以下の登場回数のk-mer部分は除去するといった具合です。
ShortReadManagerをお使いください。FASTQファイルを指定して、パラメーターをセットして実行すれば、データサイズによりますが10分から30分程度で処理が終わります。CPUパワーとメモリをかなり使用します.....
処理は2段階に分かれています。
最初の段階はcount k-merボタン(filltering based on k-mer frequencyタブ内にあります)により実行され、各k-merの出現回数を調べます。この出現回数データはアプリの内部に保持されています。
2段階目は、同じタブ内のexport curated readボタンより実行します。1段階目が終了した後で、2段階目で処理するファイルを変更することもできます。出現回数データは内部的に保持され続けます。
【手順1】setting画面で、「select read files」ボタンからfastqファイル、multi FASTAファイルを選択します(複数可)。ファイル形式は"auto detect"が良いでしょう。
【手順2】setting画面で、出力先フォルダを選択します。また、出力ファイル名のprefixを決めてください(outなど)。
【手順3】setting画面で、解析パラメーターをセットします。kの大きさ、score limit (このスコアを超える部分からk-merを数え上げます)、length limit (リード中のスコアを超える部分がこの長さを超える部分についてのみk-merを数え上げます)の値を設定します。k=21, score limit=20, length limit = 50などと設定します。
max thread、batch size、hash table sizeは、実行速度に関係するパラメーターですが、経験的に設定する必要があります。max threadは16程度を指定してみてください(分散処理を行っていますが、分散数を増やしすぎると却って速度が低下しますのでご注意ください)。batchサイズは、1000から10万程度を指定してみてください。hash table sizeは2の24乗をお勧めします。
【手順4】「Short Read Manager」ウィンドウを開き、「filtering based on k-mer frequency」タブを選びます。実行条件が表示されていますので、確認の上「count k-mer」ボタンを押してください。データサイズにもよりますが、5分から30分ほどお待ちください。進行状況は画面左上に表示されます。
【手順5】出力されたファイルをテキストエディットなどで確認してください。このデータをもとにグラフを描くなどし、何回以下のk-mer部分を除くか決めてください。
【手順6】k-mer の範囲を指定します(「filtering based on k-mer frequency」タブ内)。Try fix errorをオンにすると、出現頻度が低いk-mer部分を、出現頻度が高いk-merとなるように塩基を修正して出力します(お勧めしない機能です)。
【手順7】同タブ内の、「export curated reads」ボタンを押します。進行状況は画面左上に表示されます。出力フォルダにデータが出力されていますのでご確認ください。
kをいくつにするか決めます。SRMでは最大で31が指定できます。kは大きすぎても小さすぎてもうまくいきません。21以上をお勧めします。
k-merを数えるときに、ある程度クオリティスコアがある程度続くところだけを数えるように指定できます。
使用するスレッド数とバッチサイズを指定します(2段階目でfastqデータを処理するときのみ有効です)。
hash table sizeはそのまま(2の24乗)でどうぞ。k-merを数えるためのテーブルの大きさです。テーブルが小さいと"衝突"が増えて速度が低下します。テーブルを大きくすると衝突が減りますが、メモリ使用量が大きくなります。搭載メモリに十分な余裕がある場合はメモリの使用状況を確認しながら、数字を大きくしてみてください。
他にも色々機能があります(詳細は準備中です)。
SRMで処理可能なものに限らず、データ処理が困難な場合、PIの方よりメールにてご相談ください。特に既存のツールなどで処理できない新しいアイデアに基づくデータ処理のコーディングに興味があります。科研費などプロジェクト分担者などへの参加依頼も歓迎します。
本ソフトウエアは無保証です。バグを見つけましたらご報告いただけますと助かります。
作者: 東北大学大学院生命科学研究科 准教授 大坪嘉行
yoshiyuki.ohtsubo.a6[アット]tohoku.ac.jp
本ソフトウエアを利用した成果を公表する場合、以下の論文を引用してください (SRMはGenoFinisherプロジェクトに属しており、この論文はGenoFinisherプロジェクトの基幹となる成果です)。
Ohtsubo, Y., F. Maruyama, H. Mitsui, Y. Nagata, and M. Tsuda: Complete genome sequence of Acidovorax sp. KKS102, a polychlorinated biphenyl-degrading strain: J. Bacteriol. 194: 6970-6971 (2012)
作者は現在、新型コロナウィルスを2分程度で検出可能な迅速検出法の構築を目指しています。これまでに新型コロナウィルスに結合するDNAアプタマーを取得し、これを複数装荷した分子を作製しているところです。このような分子はコロナルウィルスが存在すると凝集体を形成させ、これを容易に検出できるものと期待しています。これまで複数の研究助成などに応募してきましたが、残念ながら研究費の獲得に至っておりません。この研究を続けるため、私の研究室へのご寄附をご検討いただければ幸いです。
寄附金は、新型コロナウィルス関連の研究ならびにソフトウエア開発などの研究教育を目的とした活動に有効に利用させていただきます(大学によって通常の研究費と同等に管理されます)。寄附金についての本学での取り扱い、税制上の優遇措置などについてはこちらをご覧ください。
ご寄附頂ける場合は、専用の様式にご記入いただき、東北大学生命科学研究科会計係までメールにてお送りください。ご検討のほど、よろしくお願いいたします。
様式はこちらからダウンロードしてください。メールアドレスはlif-kaik[at]grp.tohoku.ac.jpです。
 大学改革の方針転換を。「REUP提案」をお読みください。
大学改革の方針転換を。「REUP提案」をお読みください。
 入試や就活で「推し研」を問いませんか?
入試や就活で「推し研」を問いませんか?
 本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
 本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
 QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
 TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
 ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
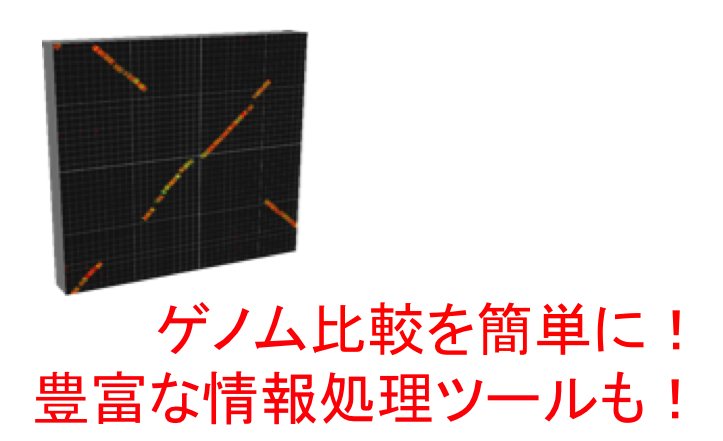 GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
 GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
 DNA配列の相補配列を計算する関数を含むエクセルシートです。
DNA配列の相補配列を計算する関数を含むエクセルシートです。
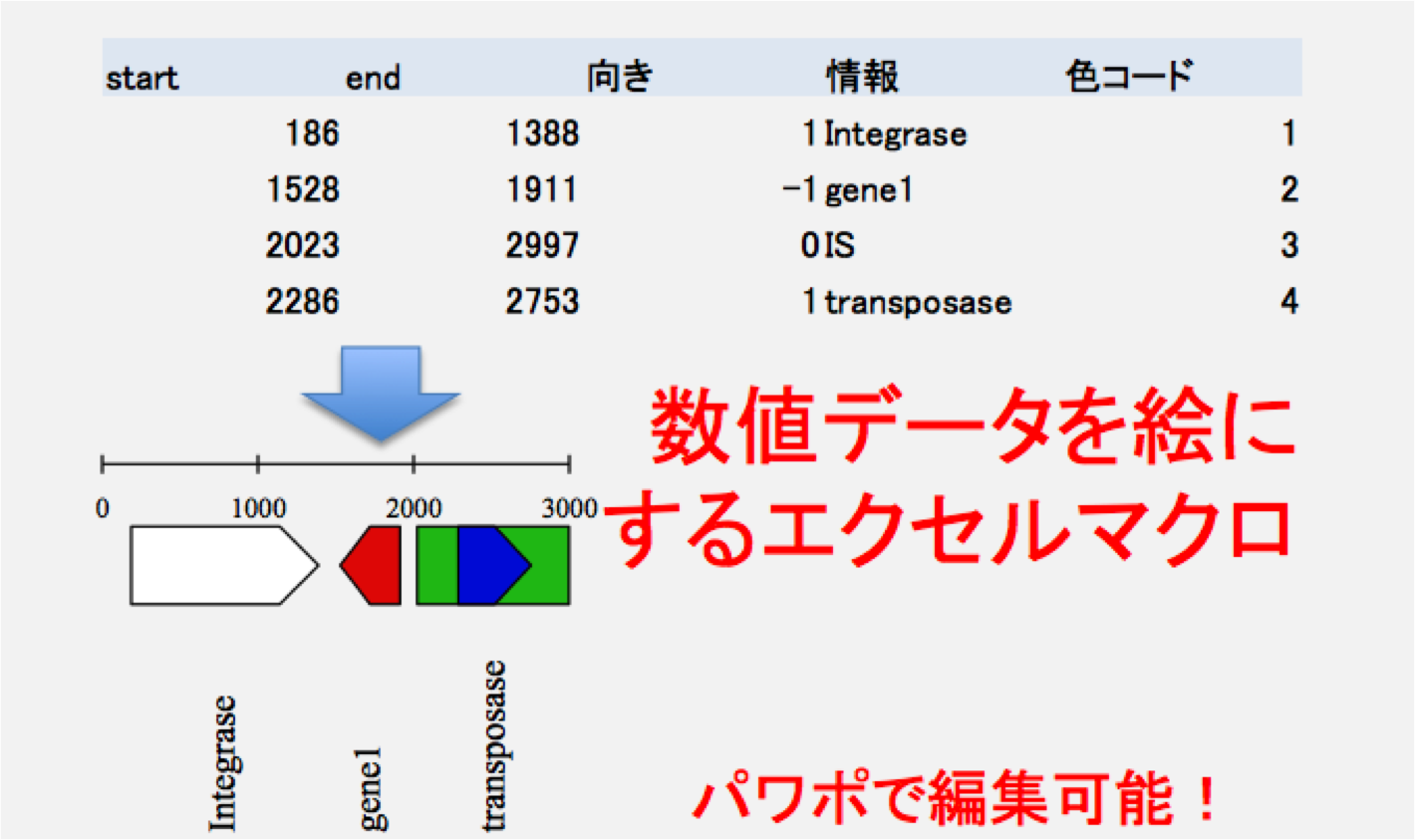 数値データを絵に変換するマクロを含むエクセルシートです。
数値データを絵に変換するマクロを含むエクセルシートです。
 数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
 大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
 DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
 例の処理を簡単に済ませるあのツールです。
例の処理を簡単に済ませるあのツールです。
 iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
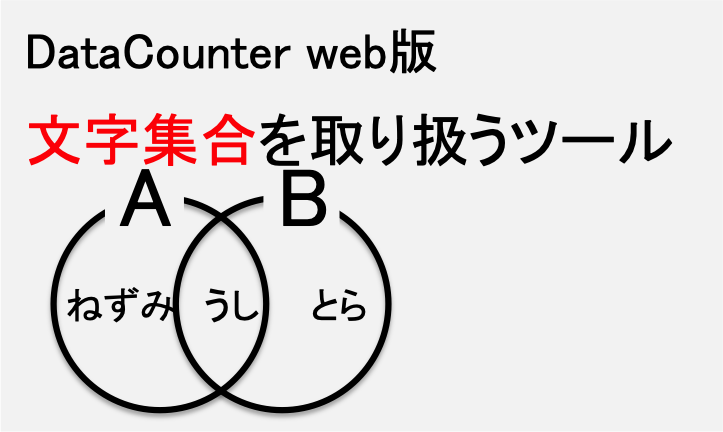 文字列集合を取り扱える便利ツールです。
文字列集合を取り扱える便利ツールです。
 GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
 文字列を取り扱える便利ツールです。
文字列を取り扱える便利ツールです。
 作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。
作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。
作者プロフィール: 環境細菌の研究を進める一方で、様々なソフトウエアを作成、公開している。