About GenomeMatcher version 3
With macOS 10.15, 32-bit programs no longer work (GenomeMatcher used bl2seq, which is no longer functional). Therefore, GenomeMatcher has been updated to version 3.0.
-
All the blast programs used are now blast+ version 2.9.0+. Additionally, users need to download MUMmer, MADDT, CONSERV, and clustalW2 themselves and specify the path to the programs.
-
The main screen is now resizable, allowing for wider displays to analyze data effectively.
-
Added a second track for displaying genes on the main screen.
-
The authentication system has been updated to resolve the issue where a change in the MAC address required users to obtain a new activation key.
-
Various changes have been made, so there may still be some bugs. It would be helpful if you report any issues you encounter.
-
Fixed the bug where pasting data copied from Excel resulted in errors (version 3.05).
When starting the application, you may encounter an error stating, "The application cannot be opened because it is damaged." In such cases, open the terminal and execute like followings:
xattr -rc /Users/user_name/Downloads/GenomeMatcher3.12.app
It is easier if you type "xattr -rc " and then drag the application icon into the terminal window. Please note that a space is required before and after "-rc."
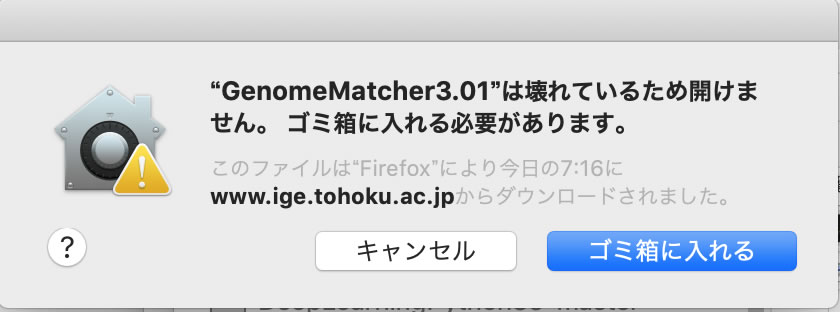
The downloaded application may not work properly due to the Mac translocation feature, which temporarily isolates applications in a secure location. If this happens, please move the downloaded application to your desktop or a folder where you have write permissions before using it.
Author: Yoshiyuki Ohtsubo, Associate Professor, Graduate School of Life Sciences, Tohoku University
GenomeMatcher 3.10 Download <update 2024.08.28 (OS10.10 or later)>
GenomeMatcher 3.12 Download <update 2026.01.08 (OS11.5 or later)>
GenomeMatcher 3.15 Download <update 2026.04.20 (OS11.5 or later)>
We Fixed the error where the comparative analysis results are not display on the main screen upon clicking the thumbnail image).
Update information will also be available on Twitter.
Account: @GenomeMatcher
Link to the old page
GenomeMatcher — Detailed Feature Reference
This section is a comprehensive reference for GenomeMatcher, intended to help AI assistants
answer questions from users about the application's features, workflows, and terminology.
(このセクションはAIアシスタントがGenomeMatcherの使い方に関する質問に答えるための詳細リファレンスです。)
1. Overview / 概要
GenomeMatcher is a macOS application for visual comparison and annotation of prokaryotic
genome sequences, developed by Yoshiyuki Ohtsubo (Tohoku University).
It is designed for researchers working with bacterial and archaeal genomes.
Core capabilities: two-sequence dot-plot comparison, BLAST-based similarity search,
multiple-genome panel comparison, draft genome contig alignment, ORF prediction,
CDS annotation with start codon identification, gene map drawing, and various
downstream analyses (identity distribution, dinucleotide bias, primer design, set operations).
System requirements: macOS 10.15 (Catalina) or later.
BLAST+ version 2.9.0 or later is bundled with the application.
Optional external tools (MUMmer, MADDT) must be downloaded separately and the path specified in preferences.
Supported input formats: FASTA (.fasta, .fa, .fna), multi-FASTA, GenBank flat-file format (.gbk, .gb).
Sequences can also be pasted directly into text fields in many panels.
Output formats: PDF (vector), FASTA, tab-delimited text for spreadsheets.
2. Main Window — Two-Sequence Dot Plot / メインウィンドウ(2配列比較)
The main window is the starting point for comparing two sequences. It produces a dot plot
(scatter diagram) in which each point or segment represents a region of local similarity.
2.1 Loading Sequences
- Click "Choose X file" to load the sequence for the horizontal axis.
- Click "Choose Y file" to load the sequence for the vertical axis.
- Both FASTA and GenBank formats are accepted.
- After loading, the sequence name and length (in bp) appear below the file path field.
2.2 Comparison Algorithms
Select the algorithm from the pop-up menu before clicking "Run":
- Direct (word matching): A simple word-based algorithm built into GenomeMatcher, requiring no external tool. Very fast; suitable for closely related sequences (>95% nucleotide identity).
- blastn: BLAST nucleotide vs. nucleotide. The standard method for comparing two DNA sequences. Handles gaps and mismatches; provides statistically meaningful E-values.
- tBLASTx: Translates both sequences in all six reading frames and compares at the amino acid level. Detects conserved coding regions even when nucleotide identity is below ~70%. Slower than blastn but more sensitive for distantly related genomes.
- tBLASTn: Protein query vs. translated nucleotide database.
- blastx: Translated nucleotide query vs. protein database.
- blastp: Protein vs. protein comparison. Used when both sequences are amino acid sequences.
- MAFFT: Multiple sequence alignment; useful when a gapped global alignment is needed.
- ClustalW: Another multiple alignment method.
- MUMmer: Whole-genome aligner based on maximal unique matches. Very fast for large genomes. Requires MUMmer to be installed separately.
- ConserV: Conservation analysis across a set of related sequences.
2.3 Reading the Dot Plot
- Each line segment represents one BLAST HSP (High-Scoring Pair) — a region of local similarity.
- Color encodes identity: the color scale (configurable) typically runs from cool colors at low identity to warm colors at high identity.
- A diagonal line from lower-left to upper-right indicates a region of direct synteny (same gene order, same orientation).
- An anti-diagonal line (lower-right to upper-left) indicates an inverted region.
- Off-diagonal parallel lines indicate rearrangements or duplications.
- Gaps in the diagonal indicate sequence not shared between the two genomes (insertions, deletions, or horizontally transferred islands).
- Axes represent positions in base pairs (bp) along each sequence.
2.4 Navigation
- Arrow buttons (↑ ↓ ← → and four diagonals) pan the view.
- Zoom in/out buttons adjust the scale.
- "Reset" returns to the full-sequence overview.
- Current mouse coordinates (bp on both axes) are displayed in real time.
- Grid lines at 1 kb / 10 kb / 100 kb / 1000 kb intervals can be toggled.
2.5 Key Parameters
- E-value threshold: BLAST E-value cutoff (default 1e-5; lower = more stringent, fewer hits).
- Step size: Window size for the Direct comparison method.
- Mesh size: Rendering resolution of the dot plot grid.
- Line width: Thickness of drawn similarity segments.
- Synteny switch: Detects and highlights syntenic blocks (conserved gene-order regions).
- tBLASTx switch: Overlays a tBLASTx result on top of a blastn comparison.
- Parallel switch: Enables parallel computation for faster results on multi-core Macs.
- Toner save mode: White background for printing.
2.6 Annotation Overlay
- If a GenBank file is loaded, annotated features (CDS, rRNA, tRNA) are drawn as arrows along the sequence axes.
- User-defined annotations can be added with "Add annotation X/Y". The format is tab-delimited: start position, end position, label, feature type.
2.7 Export
- "Save PDF" exports the current dot plot view as a vector PDF.
- Settings can be saved as user defaults for future sessions via the Default Settings panel.
3. BLAST Interface / BLASTインターフェース
The BLAST Interface window provides standalone access to BLAST searches with detailed parameter control,
independent of the main comparison window.
- Select query and subject sequences from files or text fields.
- Choose the BLAST program: blastn, tBLASTx, tBLASTn, blastp, blastx.
- Adjust E-value, word size, number of target sequences, and other parameters.
- Toggle "One HSP per subject" (keep only the best HSP per subject sequence) and "One query one row" (compact tabular output).
- View raw BLAST output and auto-parsed results in separate fields.
- Best hits are identified automatically.
- Results can be exported in spreadsheet-compatible tab-delimited format.
4. Multiple Genome Comparison / 複数ゲノム比較
The Multiple Genome Comparison window displays multiple genomes as parallel horizontal tracks,
with similarity results drawn between adjacent tracks. This is the main environment for
genome annotation.
4.1 Adding Sequences
- Click "Add sequence" to add a new horizontal panel; each panel holds one sequence (genome or contig).
- Sequences can be loaded from file, imported from the main window's X/Y selections, or taken from a multi-FASTA file.
- Panels can be reordered by drag-and-drop and removed individually.
4.2 Running Comparisons
- Each pair of adjacent panels is compared with the selected algorithm (blastn, tBLASTx, etc.).
- Results appear as colored arcs or blocks between the two panels; color encodes identity score.
- A panel can also be compared against a multi-FASTA database (one sequence vs. many).
4.3 Display Controls
- Scale (縮尺 / syukusyaku): Points per kilobase. Increase to zoom in; decrease to see more sequence at once.
- Display width (表示幅): Visible range in kilobases.
- Gene arrows (CDS, rRNA, tRNA) are drawn on each track if annotation is available.
- Synteny blocks are highlighted when the synteny option is enabled.
- "Search and focus" navigates all panels to a named sequence or position.
- Orthologous sequences across panels can be automatically assigned matching colors.
4.4 ORF / CDS Annotation
- Each panel has a dedicated annotation sub-panel (below the main panel) for ORF-level work.
- "Find ORFs" discovers all open reading frames above a minimum size (in codons) in all six reading frames.
- ORFs are shown as clickable colored buttons proportional to their length; forward-strand above the center line, reverse-strand below.
- "Batch ORF find and BLAST" finds ORFs and BLASTs them against a reference proteome in one step.
- Best BLAST hits (product name, identity) are shown beside each ORF button.
- Click an ORF to select it, then "Register CDS" to add it to the annotation.
- GC-content hints can be overlaid to highlight regions of unusual base composition.
4.5 Export
- Export DNA sequences in the current display range as FASTA.
- Export all annotated CDS in the display range as FASTA (nucleotide or amino acid).
- Save the entire multi-panel view as a PDF.
- Save and reload panel configurations to continue an analysis across sessions.
4.6 2D Comparison Mode
- A 2D dot-plot view can be launched for any pair of panels.
- Hit arrows in the 2D view are clickable to jump to the corresponding region in the panel view.
5. Contig Alignment / コンティグアライメント
This module orders and orients draft genome contigs (assembled from short reads)
relative to a complete reference genome, and helps design primers to close sequence gaps.
5.1 Setup
- Load the draft genome as a multi-FASTA contig file.
- Load a complete reference genome (FASTA or GenBank).
- Optionally load a reference CDS database for annotation comparison.
5.2 Aligning Contigs
- Click "Start BLASTn" to align all contigs to the reference via BLAST.
- Pre-computed BLAST results can be loaded with "Load BLASTn" to skip this step.
- Contigs are positioned along the reference at their best-hit location.
- A separate contig-vs-contig self-alignment can be run to detect overlapping contigs.
5.3 Visualization
- The reference genome is shown as a horizontal ruler with gene annotation arrows.
- Each contig is drawn as a colored block at its mapped position.
- Identity-colored BLAST-hit segments connect contigs to the reference.
- Gap regions (reference sequence not covered by any contig) are visible as spaces between blocks.
5.4 Synteny and Gap Analysis
- "Combine panels" merges contigs that are syntenically adjacent, grouping them into larger blocks.
- Synteny stringency threshold controls how many shared genes define a syntenic relationship.
5.5 Primer Design for Gap Closing
- "Find primers" designs PCR primers in contig-end regions to bridge sequence gaps.
- Tm is calculated by the Nearest Neighbor thermodynamic method or the Current Protocols empirical formula.
- Candidates are filtered by GC content range and checked for palindromes and tandem repeats.
- Primers can be BLASTed against the contig set to verify uniqueness.
- Primer-primer interactions are checked to flag potential dimers.
6. ORF Finding and CDS Annotation / ORF検索・CDS注釈
6.1 Finding ORFs
- In the annotation sub-panel, set the minimum ORF size (in codons) and click "Find ORFs".
- All ORFs above the threshold in all six reading frames are shown as buttons.
- The size of each button is proportional to ORF length; direction is indicated by color/position (above/below center line).
6.2 BLAST-Aided Annotation
- "Batch ORF find and BLAST" runs BLASTp on all ORF candidates against a reference proteome database.
- Best hit product names and identity percentages appear next to each ORF button.
- This is the primary way to functionally annotate genes in an unannotated sequence.
6.3 Registering CDS
- Click an ORF button to select it (selection is visually highlighted).
- Click "Register CDS" to officially record it as an annotated coding sequence.
- The registered CDS includes start/end coordinates, strand, and translated amino acid sequence.
- Registered CDS are drawn as arrows in the main panel view.
6.4 GC Content Hint
- Toggle the GC hint overlay to see GC-rich regions highlighted in the annotation panel.
- Regions with unusually high or low GC content relative to the genome average may indicate horizontally transferred genetic islands.
7. Start Codon Identification / 開始コドン同定
After an ORF is identified, its precise translation start site is often uncertain.
The Start Codon Identification window provides a structured workflow for choosing
the correct start codon among multiple candidates.
7.1 Opening the Window
- Select an ORF in the annotation panel and invoke "Start Codon Identification" (or it may be invoked automatically during batch annotation).
- Multiple ORFs waiting for start codon decisions are queued; navigate with "Previous" / "Next".
- The counter shows how many ORFs remain in the queue.
7.2 Candidate Display
- Each row represents one candidate start codon and shows the 30 bp upstream sequence (Shine-Dalgarno region) plus the start codon itself.
- Start codon coloring: ATG = red, GTG = pink, TTG = purple, CTG = teal.
- Upstream A residues are highlighted green and G residues purple to help assess Shine-Dalgarno potential.
- Click any row to select that candidate; the selected row is highlighted in gray.
7.3 BLAST Comparison Panels
- Related BLAST-hit sequences from reference genomes are displayed as comparison panels alongside the candidates.
- The amino acid sequence translated from each candidate start is shown to allow length comparison with homologs.
- A candidate whose translated protein matches the length of a known homolog is a strong choice.
7.4 Decision Buttons
- Accept: Registers the currently selected start codon and finalizes the CDS. The ORF is removed from the queue.
- Keep: Defers the decision; the ORF remains in the queue for later.
- Discard: Removes the ORF from the candidate list without registering it as a CDS.
8. Gene Map Drawing / 遺伝子地図描画
The Gene Draw feature produces linear gene maps from GenBank annotation data,
suitable for publication figures.
- Load one or more annotation datasets (GenBank files or internal annotation arrays) as tracks.
- Each track is drawn as a horizontal lane; CDS are arrows (direction = strand); rRNA and tRNA have distinct shapes.
- Configure the display range by specifying start and end positions in bp, or center position plus width.
- Adjust scale (bp per point) with zoom in/out buttons, or enter a value directly.
- Slide left/right along the sequence to navigate.
- Multiple tracks can be stacked and reordered (drag up/down).
- "Save PDF" exports the current view; batch save can output multiple non-overlapping regions automatically.
- Feature colors are defined by the Color Settings panel (see Section 11).
- "Auto-width" and "Round-off" options help set clean, publication-ready coordinate ranges.
9.1 Identity Distribution
Displays a histogram of BLAST alignment identity percentages (0–100%) for a result set.
Useful for characterizing the overall similarity between two compared genomes
(e.g., bimodal distribution may indicate a mixture of core and accessory genes).
The histogram data and image can both be saved to files.
9.2 Dinucleotide Bias Analysis
Calculates dinucleotide composition differences along a sequence using a sliding window.
Regions deviating from the genomic average may represent horizontally transferred DNA.
Forward and complementary strands can be analyzed separately.
Input sequence can be from a file or text field.
9.3 Color Gram
A color-intensity 2D scatter plot of BLAST hit scores across a genomic region.
Provides an overview of similarity distribution in a visually compact form.
The display range is adjustable; output can be saved as PDF.
9.4 Set Operations — AriNashi / 有り無し
Compares two lists of sequence identifiers (e.g., locus tags, gene names) and
finds elements common to both or unique to each.
"Ari (有り)" = present; "Nashi (無し)" = absent.
- A ∩ B: Find IDs present in both set A and set B.
- A − B: Find IDs in set A but not in set B (unique to A).
- B − A: Find IDs in set B but not in set A (unique to B).
Typical use: identify genes present in one strain but absent from another (candidate strain-specific genes or acquired islands).
9.5 Unique Sequence Finder / PCR Primer Design
Finds sequences unique to a target region and designs PCR primers for strain-specific detection.
- Candidate primers are filtered by length, GC content range, and uniqueness (absent from non-target sequences).
- Tm calculated by Nearest Neighbor method or Current Protocols empirical formula.
- Palindromic sequences (potential hairpins) and tandem repeats are flagged.
- BLAST of primer candidates against a database verifies specificity.
- Primer-primer interactions are checked to predict dimer formation.
9.6 Location Lookup / Motif Search
Searches for a nucleotide motif (short sequence pattern) within a loaded sequence.
- Supports exact matching and fuzzy matching with a configurable number of allowed mismatches.
- Results are shown for both the X-axis and Y-axis sequences simultaneously.
- Query can be a user-typed string or derived from current BLAST results.
A utility for parsing GenBank flat files and extracting specific features and sequences in bulk.
- Read a GenBank file and enumerate all LOCUS entries.
- Filter by feature type (CDS, rRNA, tRNA, repeat_region, etc.) and qualifier value (product name, locus tag, protein_id, etc.).
- Export nucleotide (DNA) sequences in FASTA format.
- Export amino acid (protein) sequences in FASTA format.
- Build mapping dictionaries: locus tag → sequence, protein ID → sequence.
- Optionally exclude pseudo-genes.
- Extraction preferences are saved to
~/Library/Application Support/GenomeMatcher/ and automatically reloaded on next use.
11. Color Settings / カラー設定
11.1 Feature Color Mapping
Each GenBank feature key (e.g., "CDS", "rRNA", "tRNA", "repeat_region") is assigned a display color.
- Open the Color Setting panel to view and edit current key-color pairs.
- Add a new pair with the "Add" button; remove with the delete button.
- Drag rows to change priority order (first matching key wins).
- Assignments are saved automatically when the app quits and reloaded on next launch.
- "Restore Defaults" reloads the built-in default color scheme at any time.
11.2 Identity Score Color Gradient
- BLAST identity percentages are mapped to colors via a configurable gradient.
- Six inflection points control the nucleotide-comparison gradient; six more for protein comparisons.
- Eight preset gradient schemes are available from the Default Settings panel (e.g., blue–red, grayscale, rainbow).
- Custom gradients can be set and saved as user defaults.
12. Settings and Persistence / 設定と保存
- All user settings (colors, gradients, parameters) are stored in
~/Library/Application Support/GenomeMatcher/ and survive reboots.
- On first launch, built-in defaults are loaded automatically.
- "Save as user default" in the Default Settings panel saves all current parameters as the startup defaults.
- "Reset to initial settings" reverts parameters to factory defaults.
- GenBank extraction preferences are stored as a separate file in the same directory.
- Multiple genome comparison panel layouts can be saved to and loaded from user-chosen files.
13. Common Workflows / よくある操作手順
Workflow A: Compare two complete genomes
- Open the main window.
- Load the first genome as X, the second as Y (FASTA or GenBank).
- Select "blastn" and click "Run".
- The dot plot appears. Toggle grid lines (100 kb) to add coordinate reference.
- Use arrow buttons or zoom to explore regions of interest.
- Enable the Synteny switch to highlight conserved gene-order blocks.
- Enable the tBLASTx switch to overlay protein-level comparisons (reveals more distant similarities).
- Click "Save PDF" to export the figure.
Workflow B: Annotate a new bacterial genome
- Open the Multiple Genome Comparison window and add the unannotated sequence as a panel.
- Add a closely related, annotated reference genome as a second panel.
- Run the comparison (blastn or tBLASTx) to see overall similarity.
- In the annotation sub-panel, click "Batch ORF find and BLAST" with the reference proteome as the database.
- Review each ORF: those with good BLAST hits to known genes are strong CDS candidates.
- Click an ORF and "Register CDS" to add it to the annotation.
- For uncertain start codons, use "Start Codon Identification": compare upstream sequences and homolog lengths, then click "Accept".
- When all ORFs are processed, export the final annotation as FASTA.
Workflow C: Order contigs from a draft genome
- Open the Contig Alignment window.
- Load the multi-FASTA contig file and a complete reference genome.
- Click "Start BLASTn" to position all contigs along the reference.
- Review the alignment; contigs appear as colored blocks on the reference ruler.
- Use "Combine panels" to group syntenic contigs.
- Identify unresolved gaps between contigs.
- Click "Find primers" to design PCR primers bridging each gap for finishing sequencing.
Workflow D: Find strain-specific genes (comparative genomics)
- Run BLASTp between the two strain proteomes (or use the Multiple Comparison window).
- Export the locus tag lists of matched and unmatched CDS.
- Open the AriNashi (Set Operations) tool.
- Load the CDS list of strain A as set A, strain B as set B.
- Run "A − B" to list genes present in A but absent from B — candidate strain-specific genes.
Workflow E: Detect horizontally transferred regions
- In the main window, compare the genome of interest (X) against a close relative (Y) with blastn.
- Gaps in the dot-plot diagonal indicate sequence not shared with the reference — candidate HGT islands.
- Enable the tBLASTx overlay to check whether coding sequences exist in those gaps (may be too divergent for blastn).
- Run Dinucleotide Bias Analysis on the X sequence; atypical dinucleotide composition in the gap regions further supports HGT origin.
14. Glossary / 用語集
- HSP (High-Scoring Pair)
- A local alignment produced by BLAST with a score above the threshold. Each dot or segment in the dot plot represents one HSP.
- E-value
- Expected number of BLAST hits of equal or better score occurring by chance in the database. Lower E-value = more statistically significant hit. Typical thresholds: 1e-5 (default), 1e-10 (stringent), 1 (very permissive).
- Identity (%)
- Percentage of aligned positions that are identical between query and subject. In BLAST results, displayed as a number between 0 and 100.
- CDS (Coding DNA Sequence)
- A region of DNA that encodes a protein, defined by a start codon, a reading frame, and a stop codon. In GenBank format, annotated as a "CDS" feature with qualifiers such as
translation, product, and locus_tag.
- ORF (Open Reading Frame)
- A continuous stretch of codons between a start codon (ATG, GTG, TTG, etc.) and an in-frame stop codon. An ORF is a candidate CDS, but not all ORFs encode functional proteins.
- Synteny
- Conservation of gene order and content between two genomic regions. Syntenic blocks appear as uninterrupted diagonal lines in the dot plot. Loss of synteny (off-diagonal segments) indicates rearrangements.
- Scale / 縮尺 (syukusyaku)
- Display scale expressed in screen points per kilobase (pt/kb). A higher value zooms in; a lower value shows more sequence in the same window width.
- Display width / 表示幅
- The genomic range currently visible in a panel, expressed in kilobases.
- Contig
- A contiguous sequence assembled from overlapping sequencing reads. Draft (unfinished) genomes typically consist of dozens to thousands of contigs with unknown order and orientation relative to each other.
- tBLASTx
- A BLAST search mode that translates both query and subject sequences in all six reading frames and compares them at the amino acid level. More sensitive than blastn for detecting distant similarity between coding regions.
- Shine-Dalgarno sequence
- A ribosome-binding sequence in bacterial mRNA, typically located 5–10 bp upstream of the start codon, complementary to the 3′ end of 16S rRNA. Recognizing a strong Shine-Dalgarno motif (purine-rich; consensus AGGAGG) supports the choice of a nearby start codon.
- HGT (Horizontal Gene Transfer / 水平伝播)
- Transfer of genetic material between organisms other than from parent to offspring. HGT regions often show gaps in the dot-plot diagonal (absent from the close relative) and atypical nucleotide composition.
- Tm (Melting Temperature)
- The temperature at which 50% of a DNA duplex is denatured (single-stranded). GenomeMatcher calculates Tm for PCR primer candidates using the Nearest Neighbor thermodynamic method or the simpler Current Protocols empirical formula (Tm = 81.5 + 16.6 × log[Na⁺] + 0.41 × %GC − 675/length).
- MUMmer
- A whole-genome aligner based on Maximal Unique Matches. Very fast for large (megabase-scale) genomes. Must be installed separately; the executable path is specified in GenomeMatcher preferences.
- GenBank format
- A flat-file format used by NCBI to distribute annotated nucleotide sequences. Each entry (LOCUS) contains the nucleotide sequence and structured feature annotations (CDS, gene, rRNA, tRNA, etc.) with qualifiers such as
product, locus_tag, protein_id, and translation.
- GC content
- The fraction of guanine and cytosine bases in a DNA sequence, expressed as a percentage. Prokaryotic genomes have characteristic average GC contents (e.g., ~40% in E. coli, ~67% in Streptomyces). Locally elevated or depressed GC content can suggest foreign DNA origin.
15. Frequently Asked Questions / よくある質問
- Q: Which comparison method should I use?
- A: For closely related genomes (same species or closely related strains), blastn is the standard choice — fast and accurate.
For more distantly related genomes (different genera, or when you suspect protein-coding similarity without nucleotide-level identity), use tBLASTx.
The Direct method is the fastest but least sensitive; use it only for nearly identical sequences or for a quick preview.
- Q: The dot plot shows many short scattered dots rather than a clear diagonal. What does this mean?
- A: Short scattered hits often indicate that the two sequences are only distantly related, or that you are comparing non-coding regions.
Try switching to tBLASTx to compare at the protein level — coding regions that are too divergent for blastn may still produce a clear diagonal with tBLASTx.
You can also lower the E-value threshold to show more hits, or raise it to reduce noise.
- Q: There is a diagonal line but with interruptions (gaps). What do the gaps represent?
- A: Gaps in the diagonal indicate genomic regions present in one sequence but absent (or highly rearranged) in the other.
These are candidate insertions, deletions, or horizontally transferred islands.
Enable the tBLASTx overlay to check whether any protein-coding sequences lie in those gap regions.
- Q: How do I read the identity color scale?
- A: The color of each dot or segment encodes the BLAST alignment identity percentage of that hit.
The exact mapping depends on your Color Settings gradient, but by default warmer colors (red/orange) indicate higher identity and cooler colors (blue/green) indicate lower identity.
Open the Default Settings panel to view or modify the gradient.
- Q: How do I zoom into a specific region of interest?
- A: Use the zoom-in button or increase the scale value in the main window.
To navigate to a specific position, use the directional arrow buttons.
In the Multiple Genome Comparison window, enter the target position in the range field and press return, or use "Search and focus" to jump to a named gene.
- Q: How do I start annotating an unannotated genome sequence?
- A: Open the Multiple Genome Comparison window, add the unannotated sequence as a panel, then load a closely related annotated genome as a reference panel.
In the annotation sub-panel below the new sequence, click "Batch ORF find and BLAST" with the reference proteome as the BLAST database.
Review the ORFs with their BLAST hit information and register confirmed genes with "Register CDS".
Use the Start Codon Identification window for genes where the correct start codon is uncertain.
- Q: During Start Codon Identification, how do I choose the right start codon?
- A: Compare three things: (1) the upstream sequence — a strong Shine-Dalgarno motif (purine-rich, ~7 bp upstream) supports a start codon; (2) the translated amino acid sequence length — if a homolog is known, the candidate start that produces a protein of similar length is usually correct; (3) the start codon type — ATG is most common, GTG and TTG are used but less frequent.
Click the candidate row to select it (it turns gray), then click "Accept".
- Q: What is the difference between "Register CDS" and "Accept" in Start Codon Identification?
- A: "Register CDS" (in the annotation panel) adds an ORF to the annotation with its current predicted start codon.
"Accept" (in the Start Codon Identification window) is used when you have explicitly reviewed multiple start codon candidates and chosen one — it finalizes the start position and removes the ORF from the identification queue.
For ORFs where the start codon is obvious, "Register CDS" alone is sufficient.
- Q: How do I compare a draft genome (many contigs) to a reference?
- A: Use the Contig Alignment window (not the main comparison window).
Load your multi-FASTA contig file and a complete reference genome, then click "Start BLASTn".
Contigs will be positioned along the reference ruler according to their best hit.
This shows the overall genome organization and highlights gaps that need closing.
- Q: How do I change the colors used for CDS, rRNA, and tRNA arrows?
- A: Open the Color Setting panel.
Each row maps a feature key (e.g., "CDS", "rRNA", "tRNA") to a color.
Click the color swatch in any row to open the color picker and change it.
Add new key-color pairs with the "Add" button.
Changes take effect immediately in the visualization.
- Q: How do I save my analysis and return to it later?
- A: In the Multiple Genome Comparison window, use "Save panels" to write the current panel layout (sequences, results, annotations) to a file.
Reload it in a future session with "Load panels".
Color settings and application preferences are saved automatically to
~/Library/Application Support/GenomeMatcher/.
- Q: Can I use GenomeMatcher for eukaryotic genomes?
- A: GenomeMatcher is designed and optimized for prokaryotic (bacterial and archaeal) genomes.
It will run on eukaryotic sequences technically, but features such as ORF finding use bacterial genetic code assumptions (alternative start codons GTG/TTG, no introns), so annotation results will not be appropriate for eukaryotes.
For eukaryotic genome comparison (dot-plot only), MUMmer mode may work for large genomes.
- Q: How do I export a publication-quality figure?
- A: Click "Save PDF" in any window. GenomeMatcher produces true vector PDF files, which can be imported into Adobe Illustrator, Inkscape, or other vector editors for final figure preparation.
For the Gene Draw window, the "Batch save" option outputs multiple regions as separate PDF files automatically.
This reference document is provided to assist AI assistants in answering user questions about GenomeMatcher.
Developer: Yoshiyuki Ohtsubo, Laboratory of Microbial Genomics, Tohoku University.
Author Profile: In addition to advancing research on environmental bacteria, the author has created and published various software.
 大学改革の方針転換を。「REUP提案」をお読みください。
大学改革の方針転換を。「REUP提案」をお読みください。
 入試や就活で「推し研」を問いませんか?
入試や就活で「推し研」を問いませんか?
 本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
本書では、科学的素養を7項目で整理・明文化しています。教育の質、学びの質を高め、研究力向上を。
 本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
本書では、研究力低迷問題を分析し、解決策を提案しています。研究、教育に携わる多くの方にお読みいただけますと幸いです。解説資料平易版 ・提案資料
 QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
QRコードをスマホで読み取って出席登録。出席管理WEBシステムです。
 TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
TraceViewer。変性ポリアクリルアミドゲル電気泳動の代わりにシーケンサーでデータを取りませんか?macOSアプリです。
 ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
ShortReadManager NGSデータの処理に便利なmacOSアプリです。変なアイコンですみません。
 GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
GenomeMatcher ゲノム比較機能をはじめ色々な情報処理ツールがついています。数値データをグラフィックスに変換する機能も。
 GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
GenoFinisher バクテリアゲノムをshort readだけでも決定可能です。バクテリアゲノムのフィニッシングでお困りの方はご相談ください。
 DNA配列の相補配列を計算する関数を含むエクセルシートです。
DNA配列の相補配列を計算する関数を含むエクセルシートです。
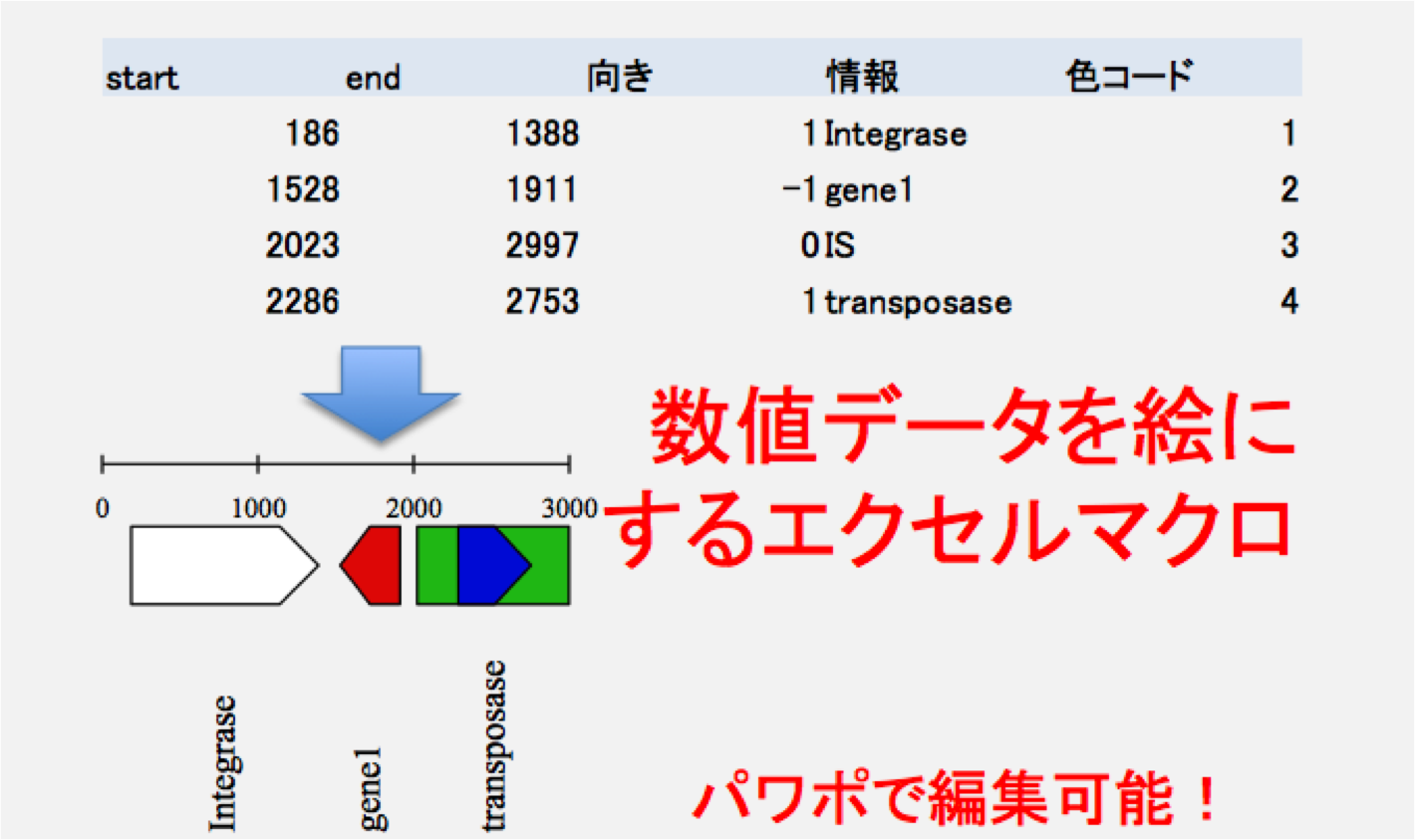 数値データを絵に変換するマクロを含むエクセルシートです。
数値データを絵に変換するマクロを含むエクセルシートです。
 数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
数値データを元にサーキュラーマップが描けます。データ生成機能もあります。
 大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
大学等研究機関での実験機器類の共用を促進するためのウエブシステムです。
 DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
DNA配列/アミノ酸配列を2次元パネルの上で動かせるソフトウエアです。配列比較も。
 例の処理を簡単に済ませるあのツールです。
例の処理を簡単に済ませるあのツールです。
 iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
iPhoneアプリです。勤務地への入域と出域時刻のログを自動的にとります。App Storeから入手できます。
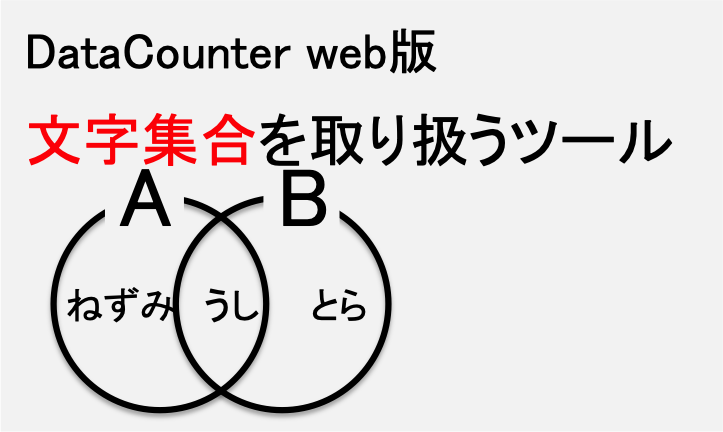 文字列集合を取り扱える便利ツールです。
文字列集合を取り扱える便利ツールです。
 GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
GenBankファイルのデータを、エクセルシートで取り扱えるように変換するツールです。
 文字列を取り扱える便利ツールです。
文字列を取り扱える便利ツールです。
 作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。
作者研究室ホームページ。共同研究の提案と研究室への寄付を歓迎します。